Some Reminiscences from an industrial spectroscopist
Bill Maddams
I became an industrial Spectroscopist by chance; or perhaps my fairy godmother decreed it, as I had an interesting and challenging career spanning forty years and could not have wished for more. At the time that I was ready to enter university, in the autumn of 1943, deferment from military service was granted only for the study of a limited number of subjects and my ambitions to pursue chemistry came to an abrupt halt. I found myself at Sheffield University on a course called Radiophysics – whose purpose, ostensibly, was to provide radar technicians. However, at that stage of World War II it was assumed, correctly, that after graduation our services in this capacity would not be required, so, in practice, I took physics as the major subject together with subsidiary electronics and mathematics. We worked four terms per year and the normally three-year course was completed in two years and three months. Hence I graduated in December 1945 at the age of nineteen.
At that stage I had no firm idea about my future; indeed, options were rather limited in the immediate post war era. In February 1946 I obtained the post of spectroscopist, working on ultra violet absorption spectroscopy, at Manchester Oil Refinery, a small concern on the western edge of the Trafford Park Industrial Estate. Although I was involved to a degree with petroleum products much of the work related to a subsidiary organisation, PetroCarbon Ltd, which had developed a cracking process that, produced mainly aromatic compounds. I worked on the quantitative analysis of mixtures of the three xylene isomers and ethylbenzene, which proved to be very good training for what I did later with infrared spectroscopy. I was also involved with the spectra of polycyclic aromatic hydrocarbons, many of which shared excellent vibrational fine structure, thus broadening my horizons.
These measurements were made with a Hilger medium quartz spectrograph and a Spekker photometer. The modus operandi of the simple double beam system was to record a series of spectra over a range of absorbance values, using a variable aperture in the reference beam, on a photographic plate. This took about twenty minutes and the plate was then developed. When dry, it was examined visually to determine the wavelength at which the intensity was equal in the beam passing through the absorbing compound and the attenuated reference beam. The usual practice was to work in absorbance increments of 0.05 up to about 1.2 and the total time required to measure a spectrum was about an hour.
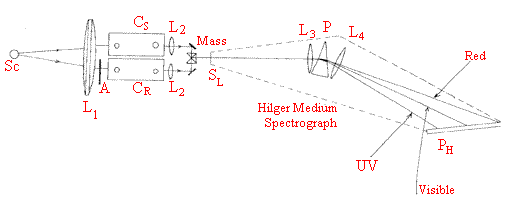
The Spekker photometer is the accessory between the Source SCand the slit SL. Radiation from the electric arc between two iron electrodes illuminates lens L1 producing parallel light, which passes through two quartz cuvettes CS (sample) and CR(reference). Transmitted radiation is collected by lenses L2 and passed to the mirror prism assembly Mass. Radiation from the sample cuvette illuminates the top half of slit SL of the Hilger Medium Quartz Spectrograph. Radiation from CR ?Bottom half of slit A is the aperture referred to in the script.
Hilger Medium Spectrograph capable of photographically displaying the line spectra of a source from the red to the UV (around 620 200nm). Range limited by the optical materials and the photographic plate sensitivity.
Light from the slit SL is collimated by lens L3 passed through prism P and re-focussed by lens L4 [old-fashioned lenses and prism are made of quartz]. The prism has dispersion maximised in the UV hence the line spectrum at the glass photographic plate PH spreads as indicated. The plate gives only a black and white image of the line spectrum.
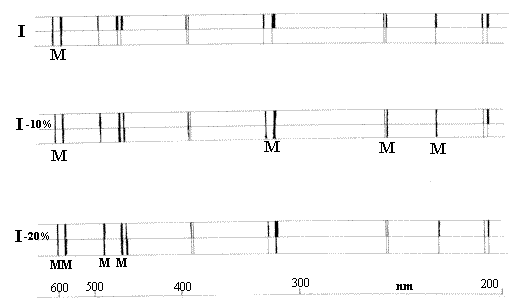
Each spectrum is represented as the Top and Bottom
half of the split – Source attenuated by Sample
Source attenuated mechanically
Top Spectrum = Sample/Reference,
2nd Spectrum Sample/Attenuated reference etc.
The poor spectroscopist looks for a match of the top and bottom spectra visually and then plots the points vs wavelength. Just to add to the little problems, the wavelength scale is incredibly non linear.
Between 1946 and 1951, I measured about three thousand spectra but felt increasingly, the need to broaden my horizons. Hence, in May 1951 I moved to the Research Department of The Distillers Co., situated in an exceedingly pleasant spot on the edge of Epsom Downs. I had been engaged to work on infrared spectroscopy in an already well-established laboratory headed by Tony Philpotts. However, as the result of staff changes I found myself involved in the determination of purities of 99 +% hydrocarbons, studies used for detailed investigations on their oxidation rates. This was done via the measurement of their freezing curves, using a thermistor as thermometer coupled to a recording Wheatstone bridge. This posed a number of problems and I was able to draw on my physics background. I also continued with some ultraviolet absorption spectroscopy using a manual instrument, and electronic determination of intensity and point by point readings but, at least, it was a step forward from photographic plates!
During this time I was rubbing shoulders with the infrared spectroscopists and learned a good deal before I became a fully-fledged member of the brotherhood. This was around the time that the first edition of Lionel Bellamy’s celebrated text on the interpretation of infrared spectra appeared and I absorbed its wisdom eagerly. Our workhorse spectrometer was a Perkin Elmer 12C single beam instrument, which although producing spectra on a chart recorder, had a considerable manual component in its operation. In order to compensate for the pseudo black-body intensity output of the source as a function wavelength, the slit width was adjusted manually, in a series of steps. Thus, commencing scanning at 15µ with a wide slit the spectrum was run as far as 14 µ, whereupon the slit width was reduced somewhat and the process was continued sequentially to the short wavelength limit. The total time required to run a spectrum was about twenty minutes.
At that time we used infrared spectroscopy extensively for the quantitative analysis of the oxidation products of cumene (isopropyl benzene) to its hydroperoxide, which was then cleaned with diluted acid to give phenol and acetone. In the oxidation process two undesirable by-products, acetophenone and phenyl dimethyl carbinol were formed and, predictably much work was done to find conditions that minimised their concentration. It proved possible to measure them at quite low levels. In the case of acetophenone (phenyl methyl ketone), the u (C=O) band was used despite interference from water vapour bands appearing in that wavelength regions because of the single beam mode of operation of the instrument. We became very proficient at the visual subtraction of this interference! Analyses of mixtures of three or four components, none of them minor, were done as necessary.
The first approach with such analyses was to set up calibration curves of absorbance as a function of concentration for each component. These sufficed if not too high a degree of precision was required. For greater accuracy matching mixtures of pure components were prepared, using micro-pipettes, whose composition approximated to the values obtained from the calibration curves. Measurements on these mixtures were then used to analyse the unknown. Although this approach was somewhat time consuming it was often possible to obtain a precision of 1% or 2% with three of four component mixtures.
A few months after I joined the team, Ray Ward arrived on the scene and his major occupation for some time was the construction of a double beam spectrometer with a photometric system. This was based on a Grubb Parsons optical unit and the challenge lay in devising a photometric system, driven by the off-balance signal between the two beams. The attenuator in the reference beam was constructed from a semi-circular metal strip into which teeth were cut so that, as it rotated, increasing attenuation was obtained. The system involved a series of pulleys beneath the optical unit and these were inter-connected by wire. The system worked well until the wire wore and broke, which happened rather too frequently for those of us who then had to tip the unit on its side and replace the wire. Surprisingly, perhaps, this in no way disturbed the optics. This spectrometer was in use for a long period of time and proved very effective.
The next addition to our armoury was a Unicam SP100 spectrometer which, by comparison, was an all singing all dancing act, with considerable flexibility in its operating conditions. Retrospectively, it was ahead of its time in at least two respects. Firstly, the analytical problems we encountered usually did not require this wide degree of flexibility. For example, we seldom needed to run high resolution vapour phase spectra. Secondly, the concepts involved in its construction were very sound, but this could not be said of all the components involved. Two problems come to mind.
It used a Golay pneumatic detector, which provided good sensitivity, but failed more frequently than we would have wished. Their replacement was rather complicated because of the construction of the optical unit. This was designed to be operated under vacuum or filled with dry air/nitrogen, and a heavy lid covering the whole unit needed to be removed. When it was replaced there was often a problem in making it airtight, as the ‘O’-ring seal did not bed down easily. By trial and error we concluded that the optimum method was to sit a young lady assistant on top of the case , turn on the vacuum pump and wait until the reduction of pressure inside provided the necessary force to ensure a good seal!
These was an appreciable amount of synthetic organic chemistry in progress, and this often produced some challenging qualitative analysis when all did not go to plan so far as the chemistry was concerned. I particularly enjoyed this type of work as it gave me the opportunity to go back to my first love, chemistry. I would try to deduce what alternative reaction might have occurred and attempt spectral interpretations on that basis. We always had excellent working relations with all of those who submitted samples and they often provided background information that was of considerable assistance. By that time gas chromatography was making an impact and trapped out GC fractions of unknowns appeared with increasing regularity. As a consequence micro infrared cells became part of our everyday routine.
In 1959 I became Section Head, Spectroscopy. The paperwork involved did not prove too much of a burden and I was still an active infrared spectroscopist. However, the scope of the Section gradually enlarged to encompass first mass spectroscopy, then x-ray fluorescence spectroscopy, plus some x-ray diffraction and finally NMR spectroscopy. Inevitably, to a degree I then became a jack of all trades, but infrared spectroscopy was not wholly a thing of the past, particularly as I became more involved with polymer characteristics.
At that time Distillers Chemicals & Plastics was well established in the PVC business and then became a licensee of the Philyro process for the manufacture of high density (linear) polyethylene. So far as this latter is concerned, the technical know-how also included an infrared method for measuring the low level of chain branching, which was rather involved and required absorbance measurements at accurately defined wave numbers. The SP100 instrument was ideal for this purpose, but not for the more direct method using a wedge of polymethylene in the reference beam. This was adjusted to eliminate the peak characteristic for methylene groups upon which the much weaker peak specific for the methyl groups of the chain branches is superimposed. The Grubb Parsons instrument proved ideal, because of the easy access to the sample compartment, which facilitated manual adjustment of the wedge. The method was automated at a later date using a Perkin-Elmer 577 spectrometer fitted with a punched tape output to record the spectral data for both sample and a polymethylene film and the subtraction process was then done on a computer.
Although PVC has been widely used for many years for a range of commercial purposes it has its limitations. Foremost among these is the way in which it darkens in use, due to the sequential elimination of HCl units, leading to the formation of conjugated polyene units, which absorb in the visible spectral region. This led to the use of a variety of stabilisers incorporated into the polymer. It had been recognised empirically that materials prepared at lower than normal polymerisation temperatures were less prone to the decomposition but, unfortunately, were too rigid and brittle for many purposes. However, the effect of polymerisation temperature on both photo and thermal degradation, a problem in sample processing when stability is required, suggested that the problem had its origin in defect structures along the chain, either rogue chemical structural units and/or faults in the conformational and configurational irregularity.
Hence, I found myself investigating this latter problem, which proved challenging but fascinating and I can only touch upon these. Quite fortuitously, the u(C-Cl) modes between about 600 and 700 cm-1 are sensitive to both the conformational and configurational structures as had already been demonstrated by Professor Krimm and workers at B.F.Goodrich. My interest lay in deciding how much quantitative information could be obtained by this approach. The inherent problem is that the various peaks in the u (C-Cl) region overlaps to varying degrees. Hence, I turned initially to the use of a Du Pont curve resolver, which is useful but has its limitations, not least because it does not provide a ‘goodness of fit’ criterion. It was also necessary to assume that the peaks are Lorentzian in shape and this, in turn, led me to measure peak shapes for a variety of simple molecules and, where deviations from the Lorentzian form occurred, find simple mathematical equations to represent them. The work was done on the PE577 spectrometer coupled to a punched tape output system, mentioned earlier.
Quite fortuitously, these studies began after The Distillers Co., sold its chemical and plastics interests to BP in 1967 and we became an out station of the main BP Research Centre at Sunbury on Thames. Although I then found myself burdened with additional paperwork, the new management system had one major advantage. Whereas the development with Distillers was done when there was any spare time, the BP system required us to devote a fixed proportion of our time to it, having first obtained approval for lines of work deemed to be of long term value. The PVC study came into this category.
Nevertheless, I realised that it required appreciably more effort than I was able to devote to it from the manpower available to me. I was fortunate in being able to pursue the matter via BP sponsored C.A.S.E. awards, with David Bower in the Physics Department at Leeds University and with Bill George, initially at Kingston Polytechnic and subsequently The Polytechnic of Wales. The Leeds work was an amalgam of our respective interests. My aim was to pursue the computer curve fitting of the PVC spectrum in greater depth and David Bower was interested in measuring molecular orientation in chain PVC specimens using Raman spectroscopy. This required a reliable quantitative separation of the overlapping peaks and, in the event, the PhD student spent three years on the problem, and obtained a great deal of useful information, both on computer curve fitting and the relative concentrations of the conformational and configurational isomers of PVC. A second student was then able to make some very useful orientation measurements.
The work done by Bill George and his students also related to these isomers, but in a different way. His interest was in making variable temperature infrared measurements to determine the energy difference between conformational isomers. He worked initially on a series of carbonyl-containing compounds and then moved to low molecular weight models for PVC, namely meso- and dl-2, 4-dichloropentone and the three configurational isomers of 2, 4, 6-tricholorheptane. Most of the work was done in solution, but some measurements were made by the matrix isolation technique, as a means for peak sharpening.
By the early 1970’s we were examining a wide range of compounds by infrared spectroscopy and were encountering an increasing number of materials, mostly solids, from which it was difficult to obtain spectra. The A.T.R technique was not always the answer. Hence, I became interested in the scope of Raman spectroscopy for dealing with such materials. Bill George, then at Kingston Polytechnic, had a JEOL Raman spectrometer and as it was not far distant from our Epsom site, Don Gerrard made occasional trips there to run exploratory spectra, which by and large, proved useful.
Around this time I read an application reporting the Raman spectrum of a thermally degraded PVC sample in which conspicuous C=C peaks appeared, from the conjugated polyene sequences that were present. What the authors had not appreciated at the time was the temperature of degradation of their sample was such that the total level of polyenes could not have been more than about 0.01%. So that the intensity of the polyene peaks must have been very high and furthermore, the sensitivity of the method offered distinct analytical potential. The authors vaguely suggested that they were observing a resonance Raman spectrum, but did not pursue the matter. This was a challenge I could not resist.
I was able to obtain PVC samples with known levels of degradation, measured via the HCl evolved, from a group working at the BP Chemicals Barry (South Wales) site, where the polymer was made and that provided the starting point for much work that proved to be both interesting and valuable. We were able to show that the spectra were indeed the result of resonance enhancement, from the presence of strong harmonic and combination peaks and other features. However, by far the most significant result was that u(C=C) varied with different exciting wavelengths, dropping in frequency as the wavelength of the exciting line increased. This is because we were exciting resonance in polyenes of different sequence lengths and u (C=C) decreases as the sequence length increases, thus providing a valuable diagnostic tool.
Soon after this initial work, the Epsom laboratories were closed and we moved to Sunbury upon Thames. This move, although logical from the company viewpoint, was not without its disadvantages. However, retrospectively, it became evident that these were strong compensating advantages. The foremost being that there was more money for equipment. Hence, it was not long before we had our own Raman spectrometer and the PVC degradation studies proceeded apace. By way of summary it suffices to say that the technique proved very valuable for assessing thermal and photodegradation and the influence of plasticisers and fillers.
We also acquired our first FTIR spectrometer soon after the amalgamation of the two laboratories and this proved exceedingly useful for both day to day analytical work and for the PVC structural studies. So far as the latter were concerned I was able to pursue other methods for studying the overlapping peak problem. Among these was second and fourth derivative spectroscopy, which leads to peak sharpening but an increase in spectral noise. Hence good quality primary data were necessary and readily obtainable by repetitive scanning. I also assessed the potentialities of Fourier self-deconvolution for peak finding, rather than for quantitative work, where considerable problems exist.
I must confess that I never ran an FTIR spectrum. During the years that preceded by retirement I was in charge of a group of about a dozen and a half people, also including specialists but administrative duties occupied a significant proportion of my time. However, I was fortunate in having capable and enthusiastic staff who would run spectra for me which, like as not, I often interpreted out of hours. I would then come back and ask for more! I progressed a long way from my initial infrared work on analysis of the oxidation products of cumene, but I always regarded myself as an applied spectroscopist. That said, I was fortunate enough to be working at a time when spectrometers and techniques advanced enormously, more or less at the same rate that increasingly challenging samples were thrust upon us. I was also fortunate in having worked with a number of spectroscopists whose interests were primarily academic, but appreciated that there is an appreciable overlap between pure and applied spectroscopy.
I could not have terminated by spectroscopic interest abruptly when I left BP and happily did not do so. However, that is another story, which does not come within the context of the present reminiscences!
REF: B. Maddams, Int. J. Vib. Spect., [www.irdg.org/ijvs] 5, 5, 3 (2001)