Raman and IR spectra of the compounds [Pl4]+[MF6]– (M = As, Sb) and [Pl4+El4–]n (E = Al, Ga, In)
Christoph Aubauer,
Thomas M. Klapötke*
and Axel Schulz
Institute of Inorganic Chemistry,
University of Munich (LMU),
Butenandtstrasse 5-13 (Haus D),
D-81377 Munich (Germany)
fax: +49-89-2180-7492
e-mail:tmk [at] cup.uni-muenchen.de
Abstract
[ PI4] +[ AsF6] – , [ PI4] +[ SbF6]– ,
[ PI4+AlI4 – ] n, [ PI4+GaI4 – ] n and the new compound [ PI4+InI4– ] n were prepared and characterised by Raman and IR spectroscopy. The vibrational spectra were compared with related compounds. Vibrational assignments for the normal modes of the PI4+ cation has been made on the basis of density functional calculations.
Introduction
The first compound containing a PI4+ cation was obtained by S. Pohl in 1983. The reaction of PI3, I2 and AlI3 in CS2 led quantitatively to the polymeric compound of the composition [PI4+AlI4 – ]n, which was structurally characterised by X-ray diffraction. [1] I. Tornieporth-Oetting and T. M. Klapötke synthesised the first isolated binary phosphorus (V) iodine cation PI4+ in 1989. The [ PI4] +[ AsF6] – salt was characterised by Raman spectroscopy. Because of the non-coordinating character of the AsF6– anion, they suggested that no significant interactions between complex cations and anions are involved for this system. [2]
Recently, M. Kaupp et al. [3] showed in a combined theoretical and experimental study, that the PI4+ cation has an extremely large negative [31]P chemical shift in the compounds [ PI4]+[ AsF6] – (-519 ppm) and [ PI4] +[ SbF6] – (-517 ppm), which is entirely due to a spin-orbit contributions from the four heavy iodine substituents, transmitted to the phosphorus nucleus by a very effective Fermi-contact mechanism. The less negative solid-state [31]P NMR chemical shifts found in the polymeric [ PI4+AlI4– ]n (-305 ppm) and
[ PI4+GaI4– ]n (-295 ppm), suggests that the P-I bond orders are reduced due to intermolecular I × × × I interaction between PI4+ cations and AlI4– anions.
Experimental Section
All the compounds reported here are moisture sensitive. Consequently, strictly anaerobic and anhydrous conditions were employed for their synthesis. All manipulations were carried out in an inert gas atmosphere (dry-box).
I2 (Merck), PI3, AlI3, GaI3 and InI3 (all Aldrich) were used as received. CFCl3 and CS2 were dried over P4O10. [ PI4] +[ SbF6] – , [ PI4+AlI4– ]n and [ PI4+GaI4– ]n were prepared according to the literature. [1, 3]The preparation of I3+AsF6– and I3+SbF6– also followed literature procedures [4,5].
Raman spectra were obtained on powdered solid samples contained in 4 mm glass capillary tubes with a Perkin Elmer 2000 NIR spectrometer fitted with a Nd-YAG laser (1064 nm) using the 180° geometry in the range at 500 – 50 cm-1. The spectra of [ PI4] +[ MF6] – (M = As, Sb) were recorded with 30 mW at -100°C using a Ventacon low temperature cell. The spectra of [ PI4+EI4– ]n (E = Al, Ga, In) were measured with
30 mW at room temperature.
IR spectra were recorded on Nujol mulls between CsI plates in the range at 800 – 200 cm-1with a Nicolet 520 FT IR spectrometer. Nujol was dried with sodium.
For the determination of decomposition points, samples were heated in sealed glass capillaries in a Büchi B450 instrument.
Preparation of [ PI4] +[ AsF6] – . In a typical reaction PI3 (0.72 g, 1.76 mmol) was reacted with I3+AsF6– (1.00 g, 1.76 mmol) in CFCl3 (15 mL) with stirring at room temperature in a two-bulbed glass vessel incorporating a coarse sintered-glass frit and a Young valve. An intense dark purple solution of iodine over a pale yellow solid was obtained. After stirring for 24 h the solution was filtered, and refiltered for several times, by condensing about half the solvent back and refiltering. Solvent and traces of remaining iodine were removed under dynamic vacuum, leaving a pale yellowish solid. Yield: 0.60 g (47 %), decomposition point 74°C.
Preparation of [ PI4+InI4– ] n. In a typical reaction PI3 (0.49 g, 1.20 mmol) was reacted with I2 (0.31 g, 1.20 mmol) and InI3 (0.60 g, 1.20 mmol) in CS2 (15 mL) with stirring at room temperature. After stirring for 24 h the solvent was removed under dynamic vacuum, leaving a black solid. Yield: 1.26 g (90 %), decomposition point 71°C.
Computational Methods. The structure and vibrational data for PI4+ were calculated by using the density functional theory with the program package Gaussian 94 (optimised d (P-I) = 2.431 Å, calculated frequency see Table 1). [6] For phosphorus a standard 6-31G(d,p) basis set was used and for I a quasi-relativistic pseudopotential (ECP46MWB) [7] and a (5s5p1d)/[3s3p1d]-DZ+P basis set. [8] The computations were carried out at the DFT level using the hybrid method B3LYP which includes a mixture of Hartree-Fock exchange with DFT exchange-correlation. Becke’s 3 parameter functional where the non-local correlation is provided by the LYP expression (Lee, Yang, Parr correlation functional) was used which is implemented in Gaussian 94. For a concise definition of the B3LYP functional see ref. [9]
| [ PI4] + [ AsF6] – |
[ PI4] + [ SbF6] – |
[PI4+ AlI4– ] n |
[ PI4+ GaI4– ] n |
[ PI4+ InI4– ] n |
Calculation ( IR) a |
Assignment |
| – | – | 380 (2) | 378 (2) | 386 (1)/376 (1) | 385 (67) | n 3 (T2), PI4+ |
| 321 (0.5) | 211 (0.5) | 194 (3.5) | n 3 (T2), EI4– | |||
| 178 (2) | 181 (3) | 152 (10) | 151 (10) | 156 (10) | 165 (0.0) | n 1 (A1), PI4+ |
| 149 (4) | 147 (3) | 134 (2) | n 1 (A1), EI4– | |||
| 82 (10) | 83 (10) | 95 (2) | 94 (2) | 99 (2) | 96 (0.1) | n 4 (T2), PI4+ |
| 71 (6) | 72 (6) | 77 (0.5) | 72 (0.5) | 88 (2) | 62 (0.0) | n 2 (E), PI4+ |
Table 1 Raman frequencies of the compounds [ PI4] +[ MF6] –(M = As, Sb) and [ PI4+EI4– ]n (E = Al, Ga, In) and the calculated frequencies for the PI4+ cation (frequencies in cm-1)
a calculated IR intensities in km/mol
Discussion of the Raman and IR spectra
Although [ PI4] +[ AsF6] – and [ PI4] +[ SbF6] – are thermally stable compounds, however, they decompose in the IR laser beam at room temperature and at low temperature, as well.
Table 1 summarises the computed and experimentally observed Raman frequencies of the PI4+ compounds. The IR frequencies of these salts are presented in Table 2. Figure 1 shows the Raman spectra of the compounds [ PI4+EI4– ]n (E = Al, Ga, In).
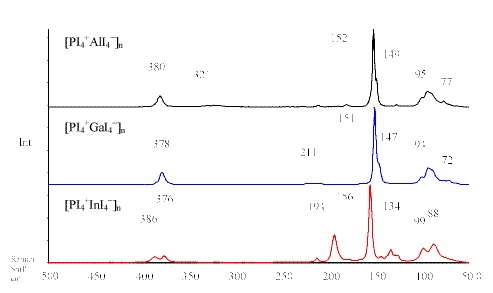
| Figure 1 Raman spectra of the compounds [ PI4+EI4– ]n (E = Al, Ga, In) |
Like in the related cations PCl4+, PBr4+ and the isoelectronic compound SiI4, possessing Tdsymmetry, there are four normal modes of vibrations expected for the PI4+ cation. The totally symmetric n 1 (A1) stretching mode can be observed with low intensity at ca. 180 cm-1 for the compounds [ PI4] +[ MF6] – (M = As, Sb). Presumably, the low intensity of the n 1 (A1) stretching mode is associated with a progressive decomposition of the compound in the laser beam. The polymeric PI4+ compounds seem to be more stable, which might be due to the strong cation × × × anion interactions. Moreover, the less intense n 1 (A1) vibration in the Raman spectra of [ PI4] +[ AsF6] – and [ PI4] +[ SbF6] – can be explained by fluorescence, often leading to wrong peak intensities in Raman spectra.
The Raman spectra of [ PI4+EI4– ]n (E = Al, Ga, In) show the most intensive peak at ca.152 cm-1 for the n 1 (A1) vibration of PI4+. This appears consistent with the suggestion, that the vibration frequencies should be at lower wavenumbers, because the P-I order in the compounds [ PI4+EI4– ]n (E = Al, Ga, In) is reduced by strong I × × × I cation × × × anion interactions, [1] whereas the PI4+ cation in [ PI4] +[ MF6] – (M = As, Sb) is almost isolated, which was shown by [31] P MAS NMR spectroscopy [3].
The sharp peaks in the Raman spectra of [ PI4] +[ AsF6] – and [ PI4] +[ SbF6] – at ca. 82 cm-1and 71 cm-1 can be assigned to the symmetric n 4 (A1) and the asymmetric
n 2 (E) deformation modes of PI4+, respectively. The Raman spectra of [ PI4+EI4– ]n (E = Al, Ga, In) show a broad tale below 100 cm-1 with two weak peaks at ca. 95 cm-1 and 77 cm-1, which can be assigned to the deformation modes n 4 (T2) and
n 2 (E) of PI4+.
The asymmetric n 3 (T2) stretching mode of the PI4+ cation can only be observed in the IR (Table 2) and Raman spectra of [ PI4+AlI4– ]n, [ PI4+GaI4– ]n and [ PI4+InI4– ]n at ca. 380 cm-1, which agree excellently with our theoretical calculation (B3LYP) for the PI4+ cation. No n 3 (T2) vibration could be observed in the IR spectra of [ PI4] +[ AsF6] – and [ PI4] +[SbF6] – , due to reaction with CsI plates.
| [ PI4] + [ AsF6] – |
[ PI4] + [ SbF6] – |
[ PI4+ AlI4– ] n |
[ PI4+ GaI4– ] n |
[ PI4+ InI4– ] n |
Assignment |
| 697 (s) | 657 (s) | n 3 (T1u), MF6– | |||
| 392 (s) | 285 (s) | n 4 (T1u), MF6– | |||
| – | – | 380 (br) | 382 (m) / 375 (m) | 386 (vs) / 375 (m) | n 3 (T2), PI4+ |
| 329 (br) | 234 (s) / 223 (vs) | n 3 (T2), EI4– |
Table 2 IR frequencies of the compounds [ PI4]+[ MF6]– (M = As, Sb) and [ PI4+EI4– ]n (E = Al, Ga, In) (frequencies in cm-1)
The presence of the anions EI4– (E = Al, Ga, In) is confirmed by the symmetric stretching mode, n 1 (A1), at 149 cm-1 ([ PI4+AlI4– ]n), 147 cm-1 ([ PI4+GaI4– ]n) and 134 cm-1
([ PI4+InI4– ]n). They are consistent with literature values (n 1 (AlI4– ): 146 cm-1;[13] n 1(GaI4– ): 145 cm-1; n 1 (InI4– ): 138 cm-1 [14]). The weak peak at 211 cm-1 (Raman) and the strong peaks at 234 and 223 cm-1 (IR) in PI4+GaI4– can be assigned to the asymmetric stretching mode, n 3 (T2), of GaI4– (n 3 (GaI4– ): 222 cm-1 [14]). The n 3 (T2) vibration of AlI4– in [ PI4+AlI4– ]n was observed at 321 cm-1 (Raman) and 329 cm-1 (IR), respectively. The peak at 194 cm-1 in the spectra of [ PI4+InI4– ]n can be assigned to the asymmetric stretching vibration, n 3 (T2), of InI4– .
The IR spectra of [ PI4]+[ MF6]– (M = As, Sb) show two expected IR active modes, n 3 (T1u) and n 3 (T1u), for an isolated MF6– anion with Oh symmetry, which are consistent with literature values. [15]
Conclusion
The Raman and IR spectra of [ PI4] +[ AsF6] – , [ PI4] +[ SbF6] – , [ PI4+AlI4– ] n,
[ PI4+GaI4– ] n and [ PI4+InI4– ] n were recorded, assigned and compared with theoretically obtained frequencies. Comparison of the spectra of the ionic with the polymeric salts show that the n 1 (A1) stretching mode is shifted to higher frequencies in the purely ionic species [PI4] +[ AsF6] – and [ PI4] +[ SbF6] – . Moreover, a new assignment of the n 1 (A1) stretching mode has been made, which was reported in a previous study at 193.5 cm-1. [2]
The Raman and IR experiments show that the isolated PI4+ cation in AsF6– and SbF6– salts are less stable than the polymeric compounds [ PI4+EI4– ]n (E = Al, Ga, In) where the cation is stabilised by strong I × × × I interactions.
Acknowledgments
We are indebted to and thank Mr. Gunnar Spieß for Raman spectroscopic measurements. Financial support by the University of Munich and the Fonds der Chemischen Industrie is gratefully acknowledged.
References
- S. Pohl, Z. Anorg. Allg. Chem. 1983, 498, 15.
- I. Tornieporth-Oetting and T. M. Klapötke, J. Chem. Soc., Chem. Commun. 1990, 132.
- M. Kaupp, Ch. Aubauer, G. Engelhardt, T. M. Klapötke and O. L. Malkina, J. Chem. Phys. 1999, 110, 3897.
- J. Passmore and P. Taylor, J. Chem. Soc., Dalton Trans. 1976, 804.
- R. J. Gillespie, M. J. Morton and J. M. Sowa, Adv. Raman Spectrosc. 1972, 1, 539.
- Gaussian 94, Revision B.2, M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. M. W. Gill, B. G. Johnson, M. A. Robb, J. R. Cheeseman, T. Keith, G. A. Peterson, J. A. Montgomery, K: Raghavachari, M. A. Al-Laham, V. G. Zakrzewski, J. A. Ortiz, J. B. Foresman, C. Y. Peng, P. Y. Ayala, W. Chen, M. W. Wong, J. L. Andres, E. S. Replogle, R. Gomperts, R. L. Martin, D. J. Fox, J. S. Binkley, D. J. Defrees, J. Baker, J. P. Stewart, M. Head-Gordon, C. Gonzales and J. A. Pople, Gaussian, Inc., Pittsburgh, 1995.
- P. Schwertfeger, M. Dolg, W. H. E. Schwarz, G. A. Bowmaker and P. D. W. Boyd, J. Chem. Phys. 1989, 91, 1762-1774.
- M. Kaupp, P. v. R. Schleyer, H. Stoll and H. Preuss, J. Am. Chem. Soc. 1991, 113,1602.
- (a) C. W. Bauschlicher and H. Partridge, Chem. Phys. Lett. 1994, 231, 277.
(b) A. D. Becke, J. Chem. Phys. 1993, 98, 5648.
(c) A. D. Becke, Phys. Rev. A 1988, 38, 3098.
(d) C. Lee, W. Yang, and R. G. Parr, Phys. Rev. B 1988, 37, 785.
(e) S. H. Vosko, L. Wilk, and M. Nusair, Can. J. Phys. 1980, 58, 1200. - P. v. Huong and P. Debat, Bull. Soc. Chim. Fr. 1972, 2631.
- M. Delahaye, P. Dhamelincourt and V. C. C. Merlin, R. Acad. Sci. Ser. B 1971, 272, 230.
- R. J. H. Clark, and T. Dines, Inorg. Chem. 1980, 19, 1681.
- G. M. Begun, C. R. Boston, G. Torsi and G. Mamantov, Inorg. Chem. 1971, 10, 886.
- L. A. Woodward and G. H. Singer, J. Chem. Soc. 1958, 716.
- T. Birchall, P. A. W. Dean, B. della Valle and R. J. Gillespie, Can. J. Chem. 1973.
Received 1st March 1999, received in revised format 29th March 1999, accepted 12th April 1999.