Using polarised vibrational spectroscopy to characterise molecular orientation in polymers: An Introduction
An Introduction – A Tutorial
Neil Everall
ICI Technology
Wilton Research Centre
PO Box 90
Wilton, Middlesbrough
Cleveland
TS90 8JE
UK
Email Neil_Everall [at] ici.com
Introduction
The physical properties of polymeric articles such as films, fibres and bottles can depend strongly upon the degree of alignment of the polymer chains. For example, properties such as tensile strength, Young’s Modulus, toughness and barrier performance all correlate with the degree of orientation. Likewise, measurements of orientation are important for testing theories of how polymer networks deform. Therefore, it is important for the polymer scientist to have access to methods for characterising orientation. There are several methods available, including X-ray diffraction, nuclear magnetic resonance (NMR), birefringence, polarised fluorescence, ultrasonic measurements, and polarised vibrational spectroscopies (IR and Raman). Of all these techniques, IR dichroism is one of the most frequently applied, since it is applicable to many polymer systems, it can be used to make measurements on the microscopic and macroscopic scales, and it can specifically measure orientation of different phases (i.e. amorphous or crystalline) in the same sample.
Given the importance of the field, this article is intended as a tutorial to introduce new practitioners to the basics of quantifying molecular orientation with vibrational spectroscopy. For the sake of simplicity we concentrate mainly on IR measurements of uniaxial (one-way drawn) systems. After discussing the basic principles, an example is given where polarised IR microscopy was used to quantify crystalline and amorphous phase orientation in drawn poly(ethylene). The theory outlined here is rigorous for uniaxial systems, but demands only basic trigonometry and calculus; hence the physical principles are not obscured by the maths which can intrude when more sophisticated treatments are applied. In a future article the treatment will be extended to include biaxially oriented systems, Raman measurements, and the experimental options for obtaining polarised IR data from real world (i.e. thick, strongly absorbing samples). For the purposes of the current article we will not dwell on how the IR spectra are obtained in cases where simple transmission measurements are not possible.
This tutorial can only serve as a brief introduction to orientation measurements. There are a number of excellent references in the literature, which cover the subject in much more detail. I would particularly recommend the books by Samuels [1] (general discussion of orientation and its measurement by a variety of techniques) and Zbinden [2] (concentrates on IR measurements, but has a large section on the different types of orientation function), although neither work discusses Raman spectroscopy. Two more modern works by acknowledged leaders in the field cover both IR and Raman measurements in detail, and also cover the rigorous analysis of biaxial orientation [3,4].
Quantifying Molecular Orientation
To see how we might quantify molecular orientation in a polymer article, first lets consider a distribution of rod-shaped molecules in the co-ordinate system defined in Figure 1. (Usually one or more of the (x,y,z) axes will be chosen to coincide with a relevant macroscopic axis such as the draw-direction of a fibre, or the in-plane optical axes of a film or bottle). Figure 1 shows the alignment of a single molecule – the orientation distribution function N(Θ,Φ,Ψ) defines the probability of the molecule having the spherical-polar co-ordinates Θ,Φ and Ψ. Obviously, to fully define the orientation one would need to measure N for all possible values of Θ,Φ and Ψ. This is in fact impossible, the best we can do in real life is to measure certain averages of the distribution function. Fortunately, these averages give useful insight into the mechanisms by which polymer chains orient on drawing, and can be correlated with end-use properties such as tensile strength, toughness, propensity to creep and so on.
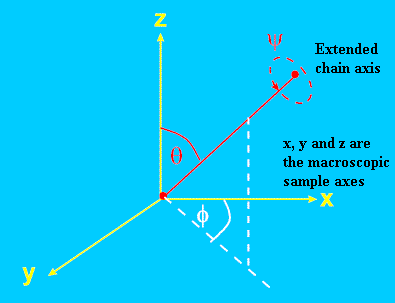
Figure 1. Definition of co-ordinate system to describe molecular orientation.
Θ andΦ are the spherical polar co-ordinates of the chain. Ψ specifies the rotation about the chain axis.
In this article we largely restrict ourselves to the case of general uniaxial orientation, i.e. systems in which only angle Θ takes non-random values. This restriction allows us to fully explore the physical basis of measuring orientation using infrared spectroscopy, but removes some of the complexity introduced with biaxial systems (where all angles Θ,Φ and Ψ must be characterised). The most obvious uniaxial example is that of a fibre, but one-way drawn films can sometimes approximate to this symmetry. To describe uniaxial orientation we only need to characterise N(Θ). We will describe two ways of doing this, one rigorous and one not, but both of which yield the same numerical result.
Orientation Averages: – P2, P4 and Fraser’s Orientation Fraction “f”
The simplest description of orientation, due to Fraser [5], makes the (unrealistic) assumption that in a uniaxially-drawn polymer article, a fraction “f” of the molecules are perfectly aligned along the draw axis (Θ=0), whereas the remainder (1-f) are isotropically oriented (Figure 2).
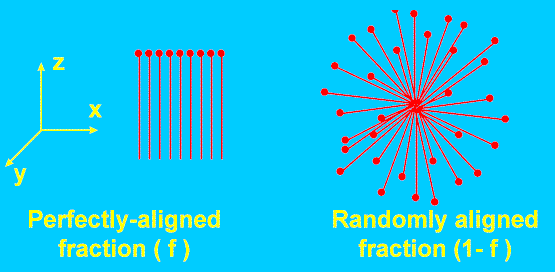
Figure 2. The simplest model of a partially oriented system assumes it is a mixture of perfectly aligned and randomly aligned molecules.
.Although this description is physically unrealistic, we find that “f” is numerically identical to the orientation parameter obtained by a rigorous analysis. One such treatment expresses the orientation distribution function (ODF) as a series expansion of Legendre polynomials [3,4]. For uniaxial orientation, with no preferred orientation about the chain director axis (i.e. Ψ random), the ODF is given by
![]()
Here Pn(cosΘ) is the nth order Legendre polynomial in cosΘ, the angular brackets denote the average value, and only terms involving even “n” are nonzero. It turns out that by using infrared spectroscopy (or any effect relying on linear dichroism) we can only measure the value of P2 for the distribution, where P2 is defined by Equation 1.
![]()
Equation 1.
P2 has the property that it varies from -0.5 for perfect alignment perpendicular to z (Θ=90), through zero for random orientation (<cos2 Θ>= 1/3 for random values of q in spherical polar co-ordinates), to unity for perfect alignment along z (Θ=0). It is identical to Hermans orientation function [6].
If one employs a two-photon technique such as polarised Raman spectroscopy or fluorescence, a second parameter, P4, can be measured (Equation 2).
![]()
Equation 2.
Figure 3 compares the variation of P2 and P4 as a function of Θ. One immediate advantage of P4 is apparent: – for highly oriented systems (Θ~0), it is more sensitive than P2 to small changes in orientation. In all cases P0 =1 so its measurement is not necessary .
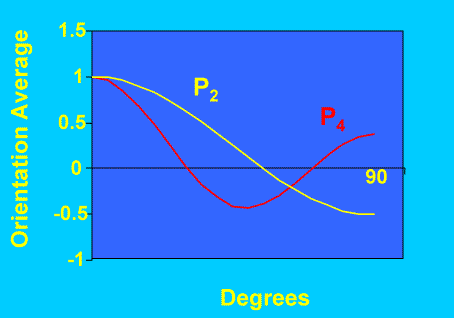
Figure 3. Variation of P2 and P4 as function of angle
Measuring Orientation with Even using Raman spectroscopy we can only actually measure the first two coefficients in the ODF (i.e. terms with n=2 and 4). To fully characterise the ODF we would need to measure Pn up to n=∞ – a time-consuming proposition even if a technique could be found! It is important to realise this fundamental limitation: – we do not measure the complete ODF using optical spectroscopy – at best we measure certain averages, up to fourth power in cosq, and we must accept that several different ODF’s could give rise to these average values. By measuring P2 we obtain the minimum level of information – with P4 we increase slightly the information content. These limitations are exemplified at the end of this article.
Measuring Orientation with Polarised Infrared Spectroscopy
Why can polarised IR spectroscopy be used to measure orientation? It is well known that molecular vibrations only give rise to infrared absorption if the molecular dipole moment changes during the vibration (in the language of quantum mechanics we say that the vibration has a nonzero transition moment m). This is a fundamental selection rule, but in itself does not guarantee that IR absorption will occur; in fact, the transition moment must have a nonzero component parallel to the electric field of the incident IR beam. The band absorbance is proportional to the square of the scalar product of the transition moment and the electric field vector, which in turn is proportional to cos2(Ω) (see Figure 4). Obviously, the absorbance is maximised at Ω=0 but is zero if Ω = 90o. This simple discussion illustrates two key points: – (a) the absorption band intensity depends fundamentally on the square cosine of an angle, and (b) we measure the orientation of oscillating dipole moments, not of molecules themselves. In a fluid, or any sample with no preferred orientation (isotropic), the band intensity will be the same irrespective of the angle of polarisation of the infrared light. If molecules are preferentially aligned in a certain direction, the band intensity will be maximised when the IR light is polarised in the same direction as the oscillating dipoles.
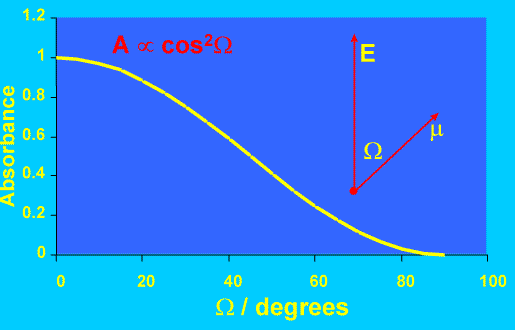
Figure 4. Variation of IR absorbance with angle between dipole transition moment and E field.
Calculating Dipole Orientation from Dichroic Ratios
To illustrate the principle of these measurements let’s first show how we can calculate Fraser’s orientation fraction f using a polarised IR experiment [1,5]. Remember that we assume the sample to consist of a fraction f of vibrational dipoles perfectly aligned parallel to the z-axis, and (1-f) randomly aligned dipoles (Figure 2). We measure two IR spectra, the first using IR light polarised parallel to the z-axis, the second with light polarised perpendicular to the axis. (For a uniaxial sample the orientation of this second axis is immaterial provided it is orthogonal to z, but for convenience lets assume we choose the x-axis). We select an absorption band, measure its peak or integrated absorbance for the two configurations (Az and Ax) and compute the dichroic ratio (D), which is defined as D = Az/Ax.
Assuming there are N vibrational dipoles in the sample, each with a transition moment of magnitude m, then the observed band intensities are given by Equations 3 and 4, where k is a constant: –
![]()
Equation 3.
![]()
Equation 4.
The first term in equation (3) is the absorbance due to the oscillating dipoles aligned parallel to z. The second term is the z contribution from the isotropically aligned dipoles – from symmetry, the projection along a given axis must be equal to 1/3 of the total transition moment for all the randomly arranged species.
The dichroic ratio is then given by: –
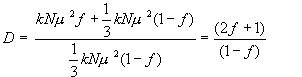
Equation 5.
Equation 5 can be rearranged to yield an expression for f in terms of D.
![]()
Equation 6.
Therefore, we can easily compute what nominal fraction of perfectly aligned dipoles would give rise to the observed dichroic ratio. Note, if the dichroic ratio is unity we obtain f=0, hence the sample is isotropic.
Effect of Angle Between Polymer Chain and Transition Dipole
So far, so good, but we are really interested in the orientation of the molecules in the sample, not the orientation of the transition dipoles. In general, the transition dipole will not lie along the extended polymer axis but will be oriented at an angle ß to it (Figure 5). Even if the molecules are perfectly aligned along z, the transition dipoles will have a component along x and y, reducing the observed dichroic ratio. We have to allow for this factor to compute the true chain orientation. In fact this is easily done using methods described by Fraser [5] and Samuels [1]. We will not repeat the derivation here but simply quote the result, which is: –
![]()
Equation 7.
Here D0 is the dichroic ratio which would be observed if the all chains were parallel to z; it can be shown that D0 = 2cot2(ß).
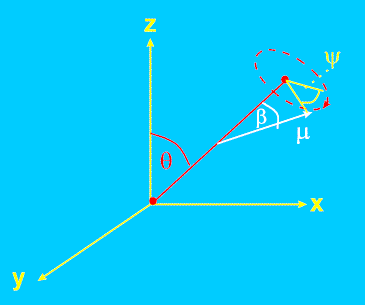
Figure 5. Orientation of dipole transition moment µ with respect to chain axis co-ordinates Θ and Ψ.
Here Ψ is the angle through which the chain rotates about its extended axis.
Rigorous Analysis – Uniaxial Alignment
A rigorous analysis of orientation has to allow for the fact that a real system does not consist of perfectly aligned molecules plus randomly aligned ones – in fact, all orientations will be observed to a greater or lesser extent depending on N(Θ,Φ,Ψ). Returning to Figure 1, (i.e. assuming for the time being thatß=0), we wish to calculate the observed dichroic ratio for an ensemble of molecules with non-random orientations. We have already seen that the absorbance observed using IR light polarised along a given axis is proportional to the square of the projected component of the transition dipole along that axis, and this in turn depends solely on ΘandΦ when ß=0. (For uniaxial orientation it depends solely on Θ, since Ψ is random for the molecular ensemble). In spherical polar co-ordinates, the z component of the transition moment µ is µcos(Θ), whereas the x component is given by µsin(Θ)cos(Ψ). The dichroic ratio is then given by Equation 8.
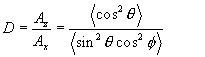
Equation 8.
The angular brackets denote the average over all molecules in the ensemble. Now, since Φ is random for uniaxial systems, it is independent of Θ, and ![]() in spherical polar co-ordinates, so we have
in spherical polar co-ordinates, so we have
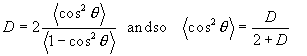
Equation 9.
Note, for random alignment, D=1 and <cos2Θ> = 1/3. In this case we cannot distinguish the situation where all molecules are in fact randomly aligned, or all molecules are aligned at ~55o to the z-axis! Both situations give the same <cos2Θ> (although the latter is unlikely)!
Combining Equations 1 and 9 we now have the required expression for P2: –
![]()
Equation 10.
Comparing this expression with Equation 6, we see that P2 and Fraser’s f are numerically equal and are easily calculated from the observed dichroic ratio.
Extension of Rigorous Uniaxial Analysis to Nonzero ß (transition dipole not parallel to chain)
The case of nonzero ß was illustrated in Figure 5. Clearly, in order to calculate the dichroic ratio, we need to compute the magnitude of the z-component of µ, and either the x or y components. This is no longer straightforward since the projection depends not only on Θ and Φ but also on ß and Ψ, the rotation about the chain axis. Papers usually quote the result (Equation 7) without showing the derivation. The result can be obtained elegantly by using the addition properties of Legendre polynomials [3,4], but this requires mathematical sophistication beyond the level of this tutorial. Fortunately, one can obtain the result by “brute force” using a straightforward, if tedious, series of co-ordinate transforms (Figure 6). They are outlined below, but the reader interested only in the practical application can skip this section.
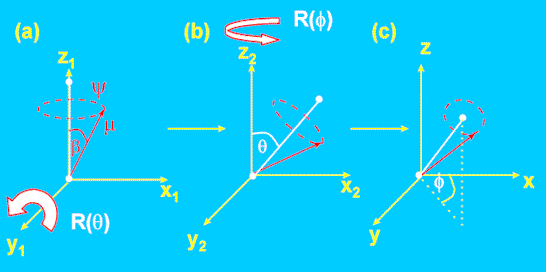
Figure 6. Co-ordinate transformations to compute dipole orientation as a function of Θ, Ψ and Φ.
First, we start with the chain aligned parallel to the z-axis. The (x,y,z) components of the dipole are: –
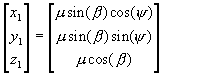
Equation 11.
We then rotate this axis system anticlockwise through an angle Θ about the y1 axis (Fig. 6a→6b), followed by a rotation anticlockwise through f about the z2 axis (6b→6c). Each rotation is characterised by a co-ordinate transformation matrix R(Θ) and R(Φ), the forms of which are derived in elementary texts [7]. The dipole co-ordinates in the resultant system are then given by Equation 12.
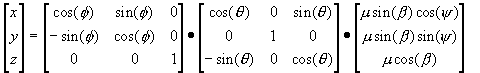
Equation 12.
The absorbance observed along the axes in the new co-ordinate system will then be proportional to the square of the x,y or z component, taking the appropriate angular averages. The reader can verify that the resultant dichroic ratio Az/Ay is given by Equation 13.
Equation 13.
To obtain this result, one needs to remember that <cosΨ> = <sinΨ> = 0 and <cos2Ψ> = <sin2Ψ> = 0.5. By substituting ß=0 the reader can verify that it reduces to Equation 9 as required. Rearranging Equation 13 to express P2 (for the chain) in terms of D we obtain Equation 14:
![]()
Equation 14.
Equation 14 is identical to Equation 7, which was derived by non-rigorous means. It tells us that the P2 value for the chain director (Θ) is given simply by the P2 value for the dipole orientation ((D-1)/(D+2)) divided by the P2 value for the dipole moment angle b. Therefore, to interpret a measured dichroic ratio we must know the value of ß=0. The extreme cases of ß=0 (parallel band, dipole parallel to chain director) and ß=90 (perpendicular band, dipole at 90 degrees to chain director) are considered in the box below.
|
Extreme Cases.
|
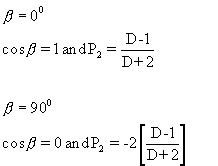
Its important to note if the dipole angle is such that cos2ß=1/3, (i.e. ß ~ 55o) then P2(cosß) = 0, and (D-1)/(D+2) = 0. This can only be the case if D=1 irrespective of the state of orientation of the sample. Vibrational bands with dipole angles close to 550 are very insensitive to orientation, the most sensitive bands have ß=0 or 90o degrees.
How do we measure ß?
There are basically three methods of obtaining ß for a particular vibration. Firstly, if we know the atomic displacements for the vibration, either through measurement (Normal Coordinate Analysis) or quantum-mechanical computation, we can calculate the direction for the dipole transition. A more common route is to measure dichroic ratios for a sample for which the orientation is already known (for example from X-ray data), and then compute ß from Equation 14. Finally, values of ß are sometimes estimated by comparison with molecules containing similar structural units, or even by “guessing” the form of a particular vibration with the aid of Group Theory. If measurements are used for comparing the same molecules under different states of orientation, errors in the value of ß are not too important. It is only when absolute values of P2 are needed that ß must be determined accurately.
Orientation Analysis using Raman Spectroscopy
Unfortunately, even for uniaxial orientation, calculating P2 and P4 values from polarised Raman data is a much more complex affair than the analogous IR calculations. This is because polarised Raman scattering is described by a tensor with up to six independent elements, as opposed to the single angle ß needed to describe the orientation of the vibrational dipole relative to the molecular axes. Even for an isotropic sample some interesting polarisation effects can arise in Raman spectroscopy, as discussed in a previous issue of IJVS [8]. In oriented systems we must calculate the transformation properties of all of the nonzero tensor elements as a function of molecular orientation – and this is a complex job [9]. It can be a major task simply to obtain the values of the tensor components themselves! For these reasons, a detailed discussion of calculating P2 and P4 from polarised Raman intensities is beyond the scope of this tutorial, and the interested reader is guided to other examples and references on the topic [3,4,10-11].
Its worth noting that in the extreme case of a single tensor component dominating the scattering, the “Fraser Fraction” approach can be used to interpret (uniaxial) Raman data. It has been shown [12] that in this case “f” is given by Equation 15
![]()
Equation 15.
Here R is the ratio of the Raman intensities Izz/Iyy , where the subscripts ij denote the macroscopic polarisation directions of the laser field (i) and the polarisation analyser (j). This is the Raman analogue of the dichroic ratio – its the ratio of the Raman intensities obtained with both laser and Raman scatter polarised first parallel, and then perpendicular to the z-axis. For an isotropic sample R=1 and f=0. For a perfectly aligned sample, R tends to infinity and f=1. However, in this case the value of “f” does not equate to the P2calculated from a rigorous analysis, it slightly overestimates the degree of orientation. Furthermore, a proper analysis also makes use of the “cross-polarisation” term Izz/Izy and so yields P4 as well as P2. Thus if all the relevant tensor components are available, a rigorous analysis should be carried out. A comparison of using the simplified and rigorous analytical approaches to study orientation of a conjugated polyene has been given elsewhere [12].
Extension to Biaxial Orientation
In the biaxial case, neither Θ nor Φ are randomly aligned, so we must calculate average orientation parameters based on both of these angles. This entails measuring two IR dichroic ratios, namely Az/Ay and Az/Ax to characterise orientation in the x, y and z directions. If the angle Ψ is random (Figure. 5), just two dichroic ratios from a single band are sufficient to characterise the distribution. However, if ß is nonzero and chains adopt preferential rotational angles about the director, it turns out that we must measure Az/Ay and Az/Ax for each of two bands with differing values of ß (ideally, a perpendicular and a parallel band) [10]. If this is done we can compute the values of <cos2εx,>, <cos2εy>and <cos2εz > where εx, εy and εz are the angles between the chain director and the x, y and z axes respectively. The details of this analysis are beyond the scope of this article but are given in detail elsewhere [10].
A Typical Application
Now let’s apply the tools developed above to the analysis of an actual oriented system. The literature contains hundreds of such examples – here we’ll discuss a very simple case, namely poly(ethylene) (PE) which has been drawn to different extents and subjected to thermal relaxation. The aim of the work was to compare orientation of the polymer under different draw and relaxation conditions in order to optimise the production process [12]. It’s a useful example since it shows how the orientation of different phases can be studied by careful choice of IR band.
The samples themselves were tubes with ca 1mm wall thickness which were cold-drawn with draw ratios 4:1 through to 7:1, and then relaxed by annealing at ~80 oC for ~30 seconds. Each tube was cut in half lengthways and an FTIR microscope (NicPlan) coupled to a Nicolet 850 FTIR bench was used to obtain spectra in transmission through the wall using IR light polarised either parallel or perpendicular to the long axis (Figure 7). Because the bands used in this study were fundamentally weak, spectra could be obtained in transmission without saturating the spectra. Strongly absorbing samples cannot be studied easily in transmission – they absorb too much radiation – and so a variety of IR techniques have been developed for such cases, including specular reflectance [13] and attenuated total reflectance [14]. These will be considered in a future article.
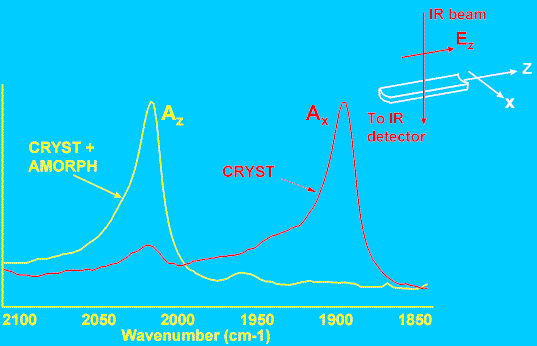
Figure 7. Polarised FTIR data from drawn PE tube.
The IR bands used for this study were located at 1894, 1078, and 2016 cm-1. The first two vibrations have been assigned to molecular conformations specific to the crystalline and amorphous phases respectively. The conformers responsible for the third band occur in both the crystalline and amorphous phases. It is therefore possible to compare orientation of the crystalline and amorphous species simultaneously – this is a major advantage over X-ray diffraction, which is sensitive primarily to crystalline-phase orientation. Figure 7 shows part of typical IR spectra obtained with the polariser aligned parallel and perpendicular to the tube long axis. It is apparent that the two bands shown here have opposite dichroism, and, following Wedgewood and Seferis [15] we assign values of ß=0 and 90o to the 2016 and 1894 cm-1 bands respectively. The dichroic ratio Az/Ax was measured for all three bands and each tube, and Equation 14 used to compute the P2 values. Figure 8 shows the results – curves (a) and (b) are the P2 values for each phase, before and after annealing, as a function of draw. These data illustrate two key points. Firstly, the crystalline phase (C) orients much more strongly than the amorphous phase (A) as a function of draw ratio. Secondly, the amorphous orientation relaxes to a much greater extent than the crystalline phase (as a proportion of the pre-annealed value). The behaviour of the conformer present in both phases (C+A) is intermediate. Data such as these are very useful in quantifying the influence of draw conditions on polymer orientation, and hence in optimising production to tailor the final properties of a polymer article.
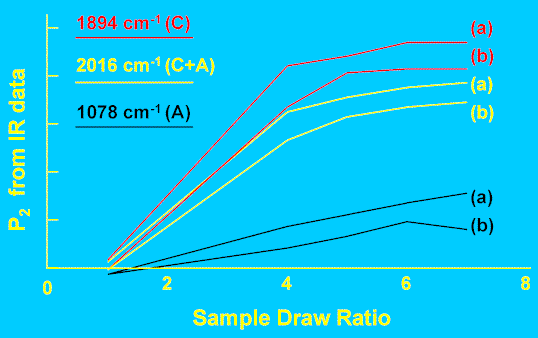
Figure 8. Phase orientation in drawn and annealed PE tubes.
(a) As drawn
(b) Post-annealed
The mechanical properties of a polymer article will be governed by both amorphous and crystalline phase orientations (perhaps to different extents), so the value of a technique that can measure both parameters is obvious. Infrared and Raman spectroscopies are not the only such options (NMR measurements are another example), but they are the most common approach. Most techniques can only measure either crystalline phase orientation (e.g. X-ray diffraction), or measure the overall orientation (crystalline plus amorphous).
What Have We Actually Measured?
At this stage its important to stress the limitations of these measurements in terms of characterising the ODF. The great problem inherent in our approach is that, at best, we measure orientation averages up to fourth power in cosΘ , we do not measure the shape of the distribution. Figure 9 illustrates this graphically – each of the distributions has the same value of <cos2Θ>,although their shapes are completely different. We cannot even calculate <Θ> without prior knowledge of the functional form of the ODF – the two distributions in Figure 9 both have <cos2Θ>~0.88, but the “exponential” curve has <Θ> of ~16o while the “delta-function” distribution has <Θ> of ~20o. This point is sometimes overlooked – a knowledge of <cos2q>or P2does not in itself give us the average angle itself, unless the shape of the ODF is known.Simultaneous measurements of P4 would narrow the range of possible distributions, but to completely specify the ODF we would need to measure order parameters up to P¥ . While we expect physical properties to correlate with P2, this correlation could break down for samples with grossly different ODF’s (e.g. comparing uniaxially and biaxially oriented systems). However, P2 and P4 values are very useful for comparing and optimising orientation in samples subjected to differing draw conditions, and can be used to investigate theoretical descriptions of polymer deformation mechanisms, provided that the overall shape of the ODF remains similar through a sample set.
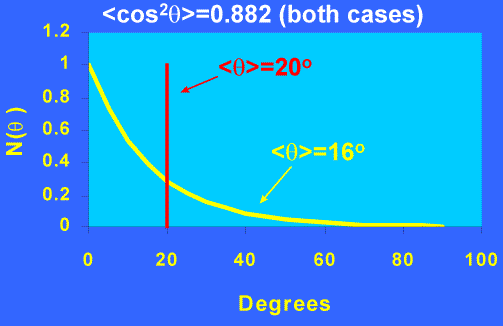
Figure 9. Comparison of two distributions with same <cos2q> but very different forms.
Conclusions
In this tutorial I have tried to give the reader sufficient grounding to enable polarised IR data from uniaxially aligned polymers to be converted into quantitative orientation results. The approach outlined is rigorous for a uniaxially aligned system yet is mathematically straightforward; a proper consideration of biaxial systems requires a more complex treatment than is appropriate here. The same is true for Raman measurements of either uniaxial or biaxial systems. Furthermore, we have not considered how one might generally obtain IR data from polymer articles – many will be too thick to analyse in transmission. For these reasons, a future tutorial will discuss experimental approaches for obtaining IR data from such articles, and the mechanics of calculating biaxial orientation functions from such data. This is important since most real-world articles (e.g. bottles, films, and plaques…) all have biaxial orientation – only fibres and tubes usually show uniaxial alignment.
Acknowledgements
The author would like to thank ICI plc for permission to publish this work. He would also like to thank John Chalmers and Peter Mills, who collaborated on the poly(ethylene) orientation work described above.
References
- R J Samuels, “Structured Polymer Properties”, J Wiley & Sons, New York, (1974).
- R Zbinden, “Infrared Spectroscopy of High Polymers”, Academic Press, New York, (1964).
- D I Bower, “Infrared dichroism, polarised fluorescence and Raman spectroscopy” in “Structure and Properties of Oriented Polymers”, Ed I M Ward, Chapman and Hall, London, (1997) pp 181-233.
- I M Ward,”Determination of Molecular Orientation by Spectroscopic Techniques” in “Advances in Polymer Science” 66, Springer-Verlag, Berlin, (1985) pp81-115.
- R D B Fraser, J Chem. Phys. 21, 1511 (1953).
- J J Hermans, P H Hermans, D Vermaas and A Weidinger, Rec. Trav. Chim. 65, 427 (1946).
- See any elementary text covering Group Theory or matrix algebra for derivation of the rotation transformation matrices. For example, J B Dence, “Mathematical Techniques in Chemistry”, Wiley Interscience, New York, (1975) pp288-293.
- A de Paepe and P J Hendra, Internet J. Vib. Spect., [www.irdg.org/ijvs], 3, 1 (1998)
- D I Bower, J. Polym. Sci., Polym. Phys. Edn. 10, 2135 (1972).
- D A Jarvis, I J Hutchinson, D I Bower and I M Ward, Polymer 21, 41 (1980).
- N J Everall, Appl. Spectrosc. 52, 1498 (1998).
- N Everall, J Chalmers and P Mills, Appl. Spectrosc. 50, 1229 (1996).
- N Everall, J Chalmers, A Local and P Mills, Vib. Spectrosc. 10, 253 (1996).
- N Everall and A Bibby, Appl. Spectrosc. 51, 1083 (1997).
- A R Wedgewood and J C Seferis, Pure and Appl. Chem. 55, 873 (1983)