Characterisation of two polymorphic forms of ranitidine-HCl
![]()
12. Characterisation of two polymorphic forms of Ranitidine-HCl
Nathan Soper*, Vivian Wu* and Thomas Rades*‡
* School of Pharmacy, University of Otago, Dunedin, New Zealand
† Department of Chemistry, University of Otago, Dunedin, New Zealand
(thomas.rades [at] stonebow.otago.ac.nz)
Abstract
The characterisation of two polymorphic forms (form I & II) of ranitidine-HCl was performed using elemental analysis, X-ray diffraction (XRD), Fourier transform-infrared spectroscopy (FTIR), diffuse reflectance infra-red Fourier transform spectroscopy (DRIFTS), Raman spectroscopy (Raman), scanning electron microscopy (SEM) and light microscopy. Differences in the diffractograms and spectra respectively of the two forms were found using XRD and vibrational spectroscopic techniques but the sample preparation involved with FTIR led to a polymorphic transition of form I to form II. Two batches of form I were investigated using the same techniques. SEM revealed that both batches showed differences in their particle shape. Differences in the signal intensity (but not in the peak position) of both batches were found using XRD. No differences between both batches of form I were detected using the vibrational spectroscopic methods. It can be concluded that XRD, DRIFTS, and Raman are suitable for qualitative analysis of ranitidine-HCl form I and II and offer potential for quantitative analysis of these polymorphs, but care must be taken using XRD for quantitative measurements as signal hight and area under the curve, amongst other factors, may also depend on the particle shape. This is not the case if vibrational spectroscopic methods are used.
Introduction
Pharmaceutical drugs and excipients can crystallise in more than one crystallographic form (polymorph, crystalline modification). Although polymorphs of a substance share the same chemical formula, the differences in the crystalline structure can affect the physicochemical parameters of the substance such as solubility, dissolution rate, density, hardness, shape, optical and electrical properties, electromagnetic spectra, diffractogram, etc. (Haleblian and McCrone 1969), which in turn can impact on important pharmaceutical properties of the drug such as bioavailability and stability of the drug as well as the formulation technology of the dosage form (Byrn et al., 1995).
The formation of different polymorphs is a controllable process within formulation. Factors such as solvent of crystallisation, rate of cooling and degree of supersaturation can effect the crystallographic form produced. Powder processing, especially pressure in the form of grinding or milling can also change the polymorphic form (Yoshiokia et al. 1994, Stagner and Guillory 1979).
The techniques used to assess polymorphism derive information from different means, and it is usually necessary to investigate polymorphism with multiple techniques. Important concerns as to which technique should be used include time and cost of the technique, the effect of sample preparation on the polymorphic form and the effect of particle shape on the analytical result.
Ranitidine-HCl (N-[2-[[[5-[(dimethylamino)methyl]-2-furanyl]methyl]thio]ethyl]-
N’-methyl-2-nitro-1,1-ethenediamine hydrochloride) is a histamine type 2 receptor antagonist used in the treatment of peptic ulcers. It has been shown that ranitidine-HCl can exist in two polymorphic modifications, forms I and II (Hohnjek et al. 1986, Carstensen and Franchini 1995) as well as in several pseudopolymorphic forms, i.e. in crystalline modifications containing solvent molecules such as ethanol or isopropanol (Madan and Kakkar 1994).
The aim of the present study was to use a range of techniques to qualitatively characterise the two polymorphs of ranitidine-HCl and to reveal possible quantitative techniques. Two different batches of form I (IA and IB) were used to investigate the influence of different particle shapes on the results of the analytical technique used.
Vibrational spectroscopic methods selected were Fourier transform infrared spectroscopy (FTIR), diffuse reflectance-infrared Fourier-transform spectroscopy (DRIFTS) and Raman spectroscopy (Raman). DRIFTS and Raman were chosen additionally to FTIR because the use of pressure in sample preparation, i.e. input of mechanical energy that may lead to a polymorphic transition, can essentially be avoided using these techniques (Coleman 1993, Gu and Jiang 1995).
Whilst FTIR, DRIFTS and Raman are techniques that detect properties associated with the molecular level, X-ray powder diffraction (XRD) is a commonly used non-spectroscopic technique to determine the polymorphic form of a substance based on differences associated with the unit cell dimensions (Brittain 1995).
Scanning electron microscopy (SEM) and light microscopy were used to determine particle morphology. These techniques do not necessarily allow the identification of different polymorphic forms of a substance, as substances with the same crystalline modification may nevertheless exhibit different crystalline habits and particle shape. On the other hand, different polymorphic forms may have the same particle shape.
Materials and Methods
Ranitidine-HCl Samples of ranitidine-HCl form II and two batches of form I (IA, IB) were supplied by Dolorgiet Pharmaceuticals, St. Augustin, Germany. The samples were stored in a desiccator at room temperature, protected from light at all times.
Elemental Analysis Elemental Analysis was performed for C, H, N and S by a gas chromatographic method using thermal conductivity with flash combustion (Carlo-Erba PA1108).
XRD This method is based on the interaction of a monochromatic x-ray beam with a crystalline substance. The different planes of atoms or molecules in a crystal act as grating for the x-rays. The conditions required for positive interference of the scattered x-ray beams to occur are given by the Bragg equation (Suryanarayanan 1995), which states that for monochromatic x-rays the diffraction angle solely depends on the crystalline spacings. The pattern provided by XRD (intensity versus scattering angle) is unique for every crystalline form of a compound. Therefore XRD is extremely useful in the identification of polymorphic forms.
XRD patterns were recorded with a Philips PW 1130 powder diffractometer (wavelength of x-rays: 0.154 nm, copper as anode material, Ni filter, acceleration voltage 40 kV, filament emission 30 mA). A Philips PW1050 goniometer was used in a step mode, with a step size of 0.04° (2θ) from 6.32° to 40.04° (2θ). Samples were compressed immediately prior to the measurement using an infrared tablet press at 2 tons. Each tablet had a weight of 1.5 g.
SEM Scanning electron microscopy was performed at 5 and 10 kV acceleration voltage using a Cambridge S360 SEM. Samples were sputtered with gold/palladium under argon vacuum resulting in an approximately 80 nm coat (Bio-rad E 5100).
FTIR FTIR was performed on a Perkin Elmer 1600 Series FT-IR using KBr discs. 1 mg of ranitidine-HCl was dispersed in 150 mg KBr. The resolution was 4cm-1 and 16 scans were averaged.
DRIFTS A Bio-rad FTS 175C dynamic alignment FT-IR spectrophotometer fitted with a Pike Technology Easidiff diffuse reflectance accessory was used. Data was captured using a PC and Win-IR software. Samples of ranitidine-HCl, form IA, IB and II were scanned 16 times. A KBr background scan was performed routinely. Ranitidine was dispersed as a 5% (w/w) mix in KBr and scanned immediately after mixing. KBr was ground for 2 minutes before geometric mixing with the ranitidine-HCl in a mortar and pestle with only light grinding was performed. The samples were then placed in the large sample cup (approx. 300 mg) using the supplied sample cup holder. A razor blade was used to smooth the sample surface. Samples were measured immediately after sample preparation.
Raman A Spectra-Physics argon laser (model 166) with a SPEX 750M spectrograph (equipped with a 1800 gratings mm-1 halographic grating) and Princeton Instruments 1152 EUV CCD detector was used. Raman spectra were calibrated using argon and neon emission lines. Calibrations were checked using known solvent spectra (Sherborne et al., 1997). Acquisition time was five minutes. The wavelength of the laser was set at 488 nm with the intensity at approximately 25 mW. Data analysis was performed using Win-IR software converting from CMSA data to Grams 32 data. Samples were analysed as supplied or geometrically mixed in appropriate ratios in an agate mortar and pestle with only light grinding. Samples were scanned within 24 hours of preparation.
Results
Results of elemental analysis are depicted in Table 1. The best fit to the results was for a ranitidine-HCl modification without ethanol, propanol or water. It can therefore be concluded that the polymorphic forms of ranitidine-HCl used in this study are true polymorphs and not pseudopolymorphic modifications.
|
Sample |
% Carbon |
% Hydrogen |
% Nitrogen |
% Sulphur |
|
IA |
44.64 |
6.53 |
16.11 |
9.11 |
|
IB |
44.71 |
6.54 |
16.15 |
9.04 |
|
II |
44.75 |
6.89 |
16.16 |
9.03 |
Table 1. Elemental analysis of ranitidine-HCl polymorphic forms
The XRD patterns of each polymorphic form of ranitidine-HCl are unique and the peak position can be used to qualitatively determine the forms of ranitidine-HCl present (Figure 1). The diffractograms are reasonably similar to those obtained by Hohnjek et al. (1986) confirming the results of the elemental analysis. For form II there are two high intensity peaks at 20.2° and 23.5° (2θ) at positions where no peaks of form I appear. These reflexes of form I are therefore suitable for a quantitative analysis of ranitidine-HCl form I and II mixtures. For form I on the other hand the peaks at 17.0°, 21.8°, and 24.9° (2θ) are characteristic.
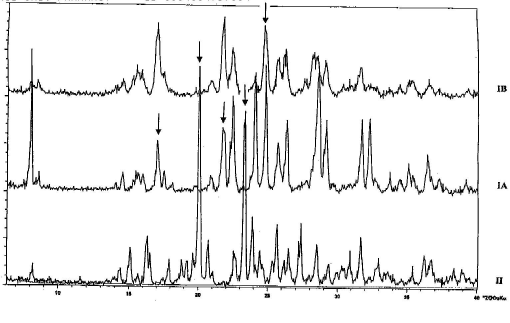
Figure 1. XRD powder diffractogram of ranitidine-HCL forms (pure powders)
The two batches of form I differ considerably in their peak intensities with form IA showing strong peaks at 8.04°, 24.2° and 32.4° (2θ) whilst form IB only shows weak peaks at the same positions. Additionally, the reflexes at 22.5°, 28.7°, and 29.3° (2θ) show a higher intensity in form IA than in form IB. A partial existence of form II in the sample of form IA or IB respectively, however, can be excluded, as in both batches of form I no reflex at 20.2° and 23.5° (2θ) can be detected.
The SEM photos show the morphology of the three ranitidine-HCl samples (plates 1-5). Form IB consists of smaller crystals that form large spherical secondary agglomerates. In contrast to form IB, form IA mainly consists of large, non-spherical, anisodiametrical particles that are not secondary agglomerates. Under pressure (as used in the sample preparation for XRD) these particles can take on a preferred orientation. The orientation of the primary particles in form IA on the other hand stays random, also after pressure was exhibited to the sample (micrographs not shown). Form II has an intermediate crystal size range and may also exhibit preferred orientation when packed under pressure.

Plate 1. Ranitidine-HCl form IA (horizontal field ˜ 1 mm)

Plate 2. Ranitidine-HCl form IA (horizontal field ˜ 125 µm)

Plate 3. Ranitidine-HCl form IB (horizontal field ˜ 6 mm)

Plate 4. Ranitidine-HCl form IB (horizontal field ˜ 80 µm)
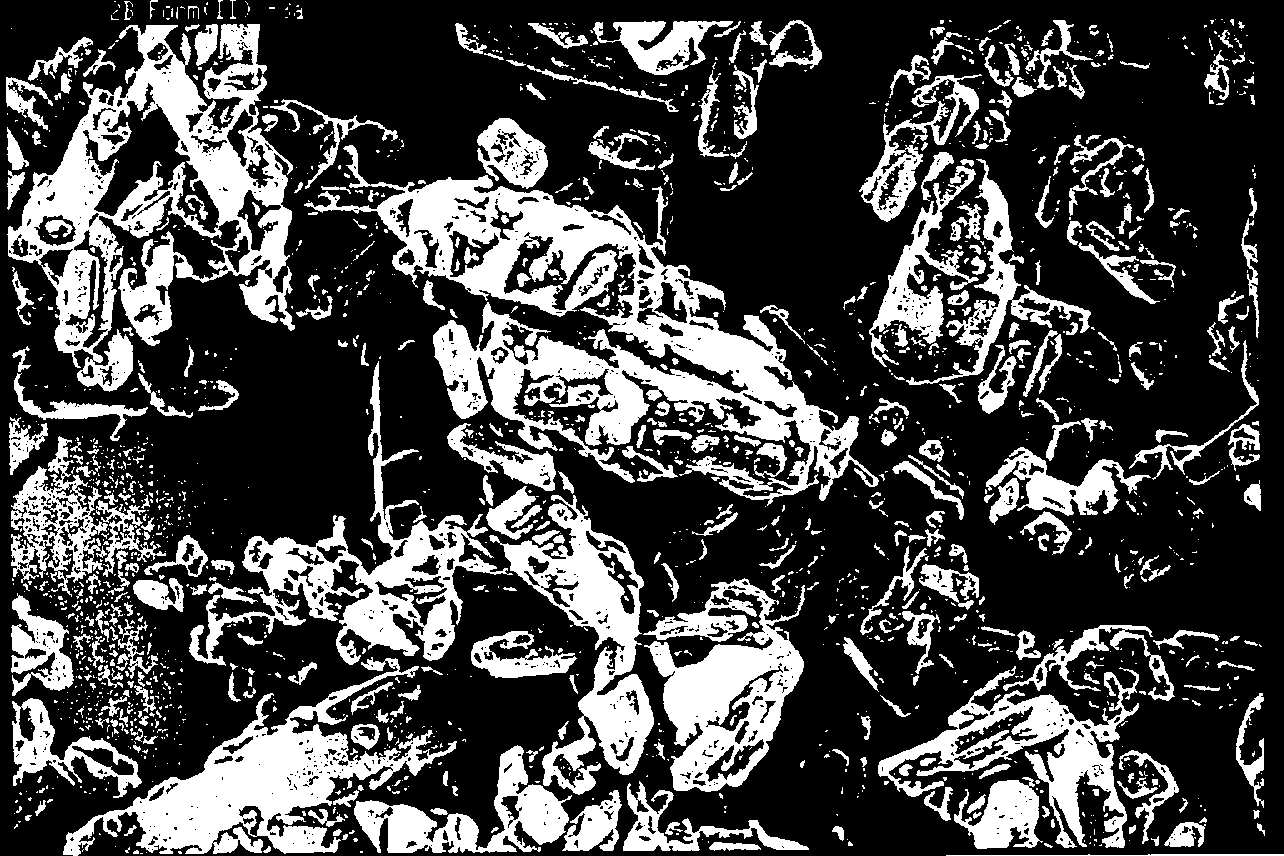
Plate 5. Ranitidine-HCl form II (horizontal field ˜ 100 µm)
The DRIFTS spectra are detailed in the figure 2. Form II can be qualitatively determined by the strong peak at 1046 cm-1. Form I can be qualitatively determined from the peak at 1551 cm-1. Forms IA and IB are an exact match when compared, evidence that they are the same polymorphic form. When in a mixture (1:1) the peaks specific for both form I and form II are clearly evident (figure 3) and are suitable for quantitative studies.
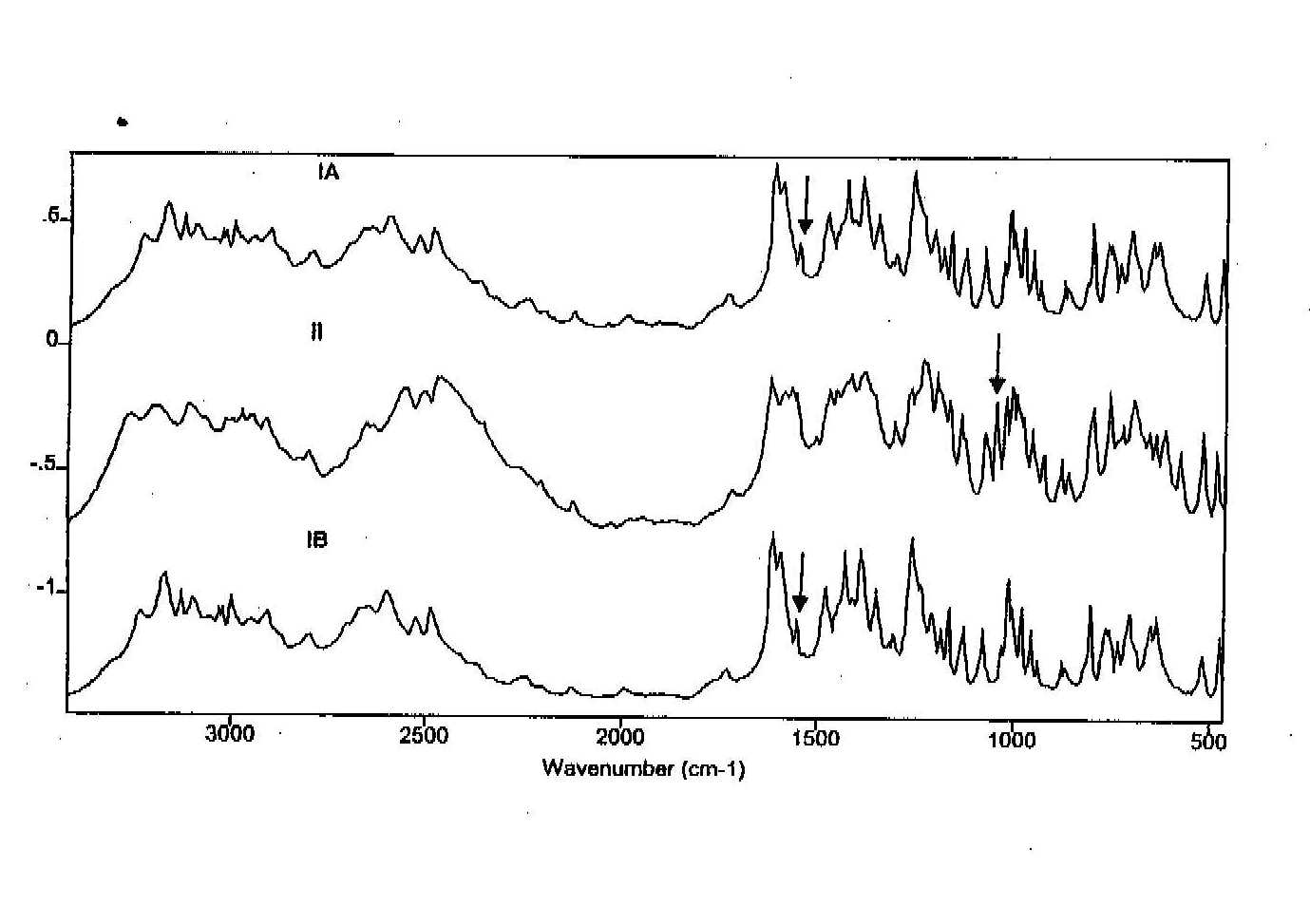
Figure 2. DRIFTS spectra of ranitidine-HCl forms (5% in KBr)
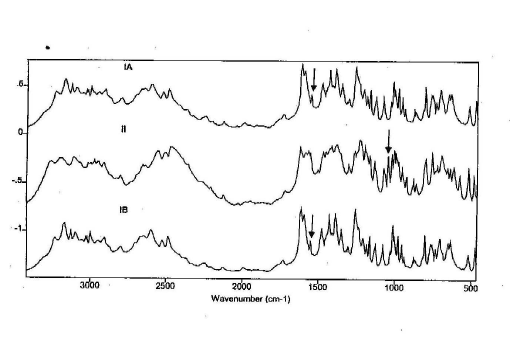
Figure 3. DRIFTS spectrum of a 1:1 mixture of ranitidine-HCl forms IB and II (5% in KBr)
FTIR results showed the transformation of form I into form II during sample preparation with peaks characteristic of form II appearing in the form I spectra after grinding (peak at 1046 cm-1) (figures 4 & 5).
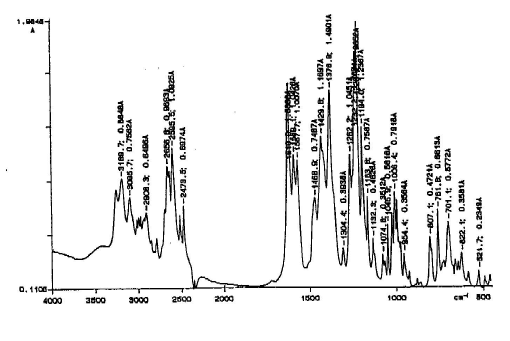
Figure 4. FTIR spectrum of ranitidine-HCL form I (KBr disc)
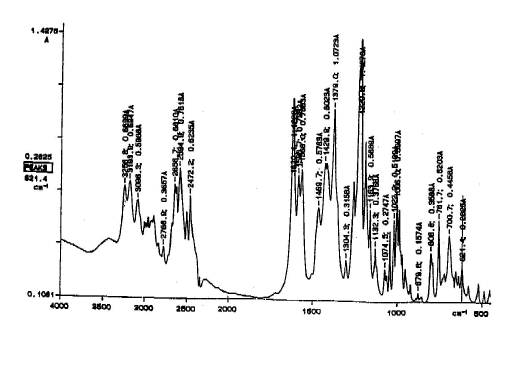
Figure 5. FTIR spectrum of ranitidine-HCL form II (KBr disc)
Also the Raman spectra of ranitidine-HCl form I and form II exhibit characteristic peaks that allow the identification of the polymorphic form (figure 6). The differing features include form II peaks at 1305.7 cm-1, 1185 cm-1, two shoulder peaks on either side of the peak at 1247 cm-1, and form I peaks at 1208 cm-1 and 1120 – 1140 cm-1. Of main interest for qualitative purposes are unique, strong intensity peaks. When the spectra were overlaid the form I peak at 1208 cm-1 and the form II peak at 1185cm-1 were judged most suitable for identification. These peaks also offer potential for a quantitative analysis of polymorphic mixtures. When in a mixture the peaks specific for both form I and form II are clearly evident and are suitable for quantitative studies. The spectra of form IA and form IB were similar enough to be considered identical.
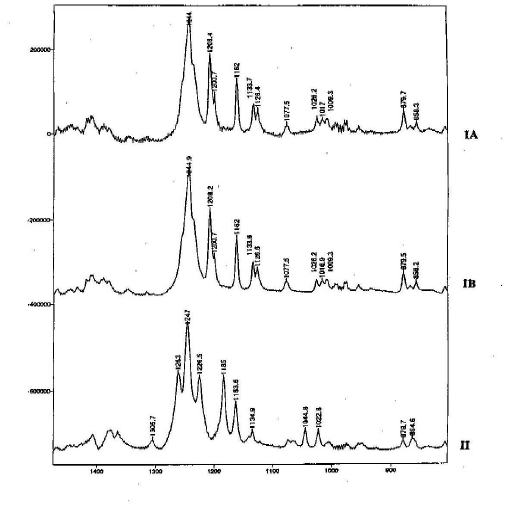
Figure 6. Raman spectra of ranitidine-HCl forms (pure powder)
Discussion
Elemental analysis determined form IA, form IB, and form II to be ranitidine-HCl without incorporation of solvents.
Differences in all measuring procedures were recorded for form I and II of ranitidine-HCl. Qualitative analysis can be carried out routinely using XRD, DRIFTS and Raman spectroscopy. However, with FTIR transformation under pressure from form I to form II occurred, hence this technique is not suitable for qualitative (and consequently also for quantitative) analysis. Photo-acoustic detection, due to the lack need for sample preparation, would also be a suitable technique for FTIR measurement.
The differences between the polymorphic forms in vibrational spectroscopic techniques are due to either differences in the dipole moment or polarity and are caused by differences in the internal structure of the polymorphs on a molecular level. The differences in the DRIFTS spectra point to minor conformational differences in hydrocarbon bending.
The differences in XRD diffractograms on the other hand are due to different crystalline spacings. Different polymorphic or pseudopolymorphic forms will exhibit peaks at different diffraction angles, i.e. will have different crystalline spacings. The peak position therefore is used to identify the polymorphic form. The peak intensity however, can vary drastically depending on the orientation of the particles in the sample holder. These textural effects are especially troublesome in goniometer measurements, as only a cut through the diffraction cone of the sample is detected (Krischner 1990). The batches of form I showed differences in the peak intensities in the XRD diffractograms. SEM investigations revealed that these differences can be attributed to different particle forms. DRIFTS and Raman spectroscopy are not sensitive to changes in the particle form of different batches of form I.
Conclusions
- Qualitative analysis of different polymorphic forms of ranitidine-HCl is possible with XRD, DRIFTS and Raman, but not with FTIR as sample preparation can lead to polymorphic changes.
- Of the qualitative techniques presented, DRIFTS and Raman are quick, simple and relatively inexpensive. XRD is also applicable to qualitative analysis but is more time consuming, especially when the whole diffractogram is recorded and the technology is often unavailable.
- In terms of a quantitative analysis of the two polymorphic forms of ranitidine-HCl, both XRD and vibrational spectroscopic techniques are potential candidates provided accurate sample preparation can be performed, which is the major prerequisite to achieve reproducible results.
References
- Brittain, H.C., Overview of physical characterisation methodology. In: Brittain, H.C. (ed.), Physical characterisation of pharmaceutical solids. pp 1-35, Marcel Dekker Inc., New York, 1995.
- Byrn, S., Pfeiffer, R., Ganey, M., Hoiberg, C. and Poochikian, G., Pharmaceutical solids; a strategic approach to regulatory considerations. Pharm. Res. 12(7), 945-954 (1995).
- Carstensen, J.T., Franchini, M.K., Isoenergetic polymorphs. Drug Development and Industrial Pharmacy, 21(5), 523 536 (1995).
- Coleman, P.B., Practical sampling techniques for IR analysis, CRC press, London, 1993.
- Gu, X.J. and Jiang, W. Characterization of polymorphic forms of fluconazole using Fourier transform Raman spectroscopy. J. Pharm. Sci. 84(12),1438-1441 (1995).
- Haleblian, J.K. and McCrone, W., Pharmaceutical applications of polymorphism. J. Pharm. Sci. 58(8) 911-929 (1969).
- Hohnjec, M., Kuftinec, J, Malnar, M. (et al.), Ranitidine. In: Florey, K (ed.), Analytical profiles of drug substances. Vol. 15, pp 533-561, Academic Press, New York (1986).
- Krischner, H. Einführung in die Röntgenfeinstruktur-Analyse. 4th ed., pp 45-48 Friedr.Vieweg und Sohn Verlagsgesellschaft, Braunscheig, 1990.
- Madan, T. and Kakkar, A.P., Preparation and characterization of ranitidine-HCl crystals. Drug Development and Industrial Pharmacy 20(9) 1571-1588 (1994).
- Sherborne, J., Scott, S. and Gordon, K., Spectrochemical studies of some ruthenium(II) complexes with polypridyl bridging ligands. Inorganic Chimica Acta. 254, 267-272 (1997).
- Stagner, W.C. and Guillory, J.K., Physical characterization of solid iopanoic acid forms. J. Pharm. Sci. 1979; 68(8): 1005-1009.
- Suryanarayanan, R., X-ray powder diffractometry. In: Brittain, H.C. (ed.), Physical characterisation of pharmaceutical solids, pp 187-221. Marcel Dekker Inc., New York. 1995.
- Yoshiokia, M., Hancock, B.C. and Zografi, G., Crystallisation of indomethacin from the amorphous state below and above its glass transition temperature. J. Pharm. Sci. (1994); 83(12): 1700-1705.
Acknowledgments
The authors wish to thank: Dolorgiet Pharmaceuticals, St. Augustin, Germany for supplying the Ranitidine-HCl, Damian Walls, Department of Geology, University of Otago, for his help with XRD and Mark Gould, Department of Anatomy and Structural Biology, for his assistance with SEM.
Received 23rd Jauary 1998, received in revised format 3rd March 1998, accepted 4th March 1998.