FT-IR: A useful analytical tool for monitoring of solid phase organic reactions
Christophe Fromont
Department of Chemistry
University of Southampton
Southampton
SO17 1BJ
U.K.
Introduction
Although solid phase chemistry has been around for a long time [1], it has seen a real expansion since the early 1990’s with the introduction of combinatorial chemistry. Several reviews [2] have been devoted to combinatorial chemistry, as well as web sites (5z.com., Combinatorial.com., warr.com.). Rapid progress in molecular biology and automated screening techniques (robots can now evaluated several hundred thousand of compounds per year) has lead to a lack of candidates for screening. Traditionally, candidates were provided either by nature [3] or by design and production by organic chemists. Products assembled by classical solution phase chemistry are often not very diverse, while on the other hand, production by nature is much more diversified but requires lengthy and sometimes difficult purifications, and finally, can lead to a complex total synthesis. The demands of high throughput screening within the pharmaceutical industry have therefore boosted recent expansions in the field of combinatorial chemistry [4].
What is combinatorial chemistry?
In traditional solution phase chemistry, two reagents A and B are reacted together to give a compound A-B. This technology is limited to the production of one compound or a small mixture of compounds per flask and purification is compulsory if an excess of reagent is used (Figure1).
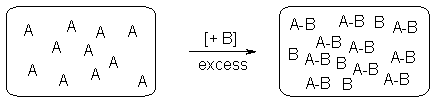
Figure 1. Classical solution phase chemistry
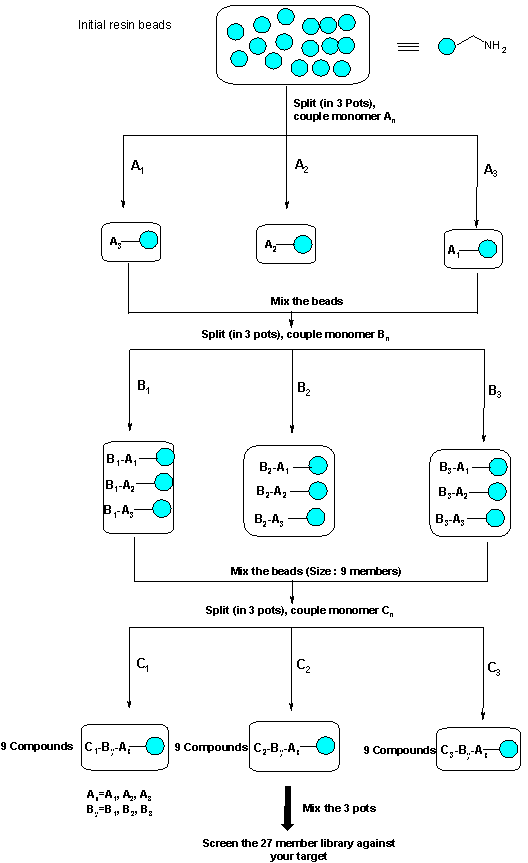
Figure 2“Split and Mix” Solid phase chemistry
The power of combinatorial chemistry lies in the use of solid supports and the so called “split and mix” strategy [5] to allow the production of several thousand of compounds in one flask, the ultimate reactor being the resin bead itself (one bead one compound)[6] (Figure 2).
Mixing and splitting ensures the statistical presence of each compound in a library. Since the reaction rate for a given compound depends on the resin [7] and on the reactants, each reaction step is carried out in different batches.
The initial batch of resin, for example, is divided into 3 batches corresponding to 3 reagents (A1, A2, A3) used for the first coupling. In each batch, the reaction is driven to completion by using an excess of reagent (easily removed by simple washing of the support), and possibly several coupling cycles. To ensure the overall representation, the batches are mixed together and split again into 3 batches for the second coupling with 3 other reagents (B1, B2, B3). The resulting mixed batch now contains 9 compounds. The sequence is repeated until the end of the solid phase synthesis.
The size of a library (a library is the set of all the compounds produce in a combinatorial synthesis) obtained will depend on the number of reactants or building blocks (linear relation) and the number of reaction steps (exponential relation). In this case, a library of 33 compounds equally represented is obtained (Figure 2).
Although solid phase synthesis has a number of advantages, new problems have risen:
- a) whereas the creation of a whole library takes only a few days, the development of a new reaction on resin can take several months,
- b) the product must be released from the resin by an extra step of cleavage,
- c) last but not least, since the resin bound intermediates can not be purified, it is crucial that the efficiency of every synthetic step must be confirmed. However, conventional analytical techniques in solution are no longer applicable. The need for reliable analytical techniques on solid phase was felt in the early seventies [8], since most organic reactions don’t go to completion. Unlike solution phase synthesis, unreacted materials bound on the resin can not be removed after each reaction step. They will accumulate or react during other steps and so will cause purity problems and decrease the yield of the final product.
Analytical techniques used to monitor and analyse the synthesis of a compound in the solid phase:
We will focus in this review on “on bead” analysis of the product, since if the reaction is not complete on the resin, lower yield and lower purity will result. In the first part, we will briefly summarise the different methods to monitor reactions on solid phase and methods to characterise the final product. In the second part, we will concentrate on infrared techniques adapted to the solid phase.
- I) Monitoring a solid phase reaction
Some other recent reviews has been published [9,10,11,12,13] , and present a good overview of different methods used to monitor the synthesis or analyse a compound resulting from solid phase synthesis.
Colorimetric tests:
The Ninhydrin (or Kaiser’s) test is a useful primary amine colour test, which has been used to quantify the loading of aminomethyl resin [14]. The Fmoc test, which consists of coupling a Fmoc protected amino acid or Fmoc chloroformate and measuring the UV absorbence of the fulvene derivative after deprotection using 20% piperidine in DMF, is also used to calculate the loading on solid phase [15]. For couplings to proline or secondary amines, bromophenol blue is used as indicator [16]. Ellman’s reagent bis (3-carboxyl-4-nitrophenyl) disulphide has been used to monitor reactions with sulfhydryl group [17].
NMR spectroscopy:
The first technique used was the “gel phase NMR”. This consists in taking a slurry of polymer bound organic substrate swelled in an appropriate solvent [18,19] and running a standard NMR. NMR Spectra of 19F, 15N, 31P, 1H and 13C have been recorded and used to monitor solid phase reactions. The main drawback of this method is the low sensitivity because of the relatively low amount of compound on the resin (usually, 100mg of resin (1.2 mmol/g) are used). For 1H NMR the broadness results in loss of information and poor structure determination. Long accumulation times or labelled carbons [20] are useful to obtain suitable signals. To circumvent this low resolution, MAS NMR (Magic Angle Spinning) has been adapted from the solid state NMR technique. In this method, a special probe is needed and the spectra is recorded from the sample still in his solid state (in fact, swollen in a minimum of solvent)[21,22] . MAS NMR leads to NMR spectra approaching the quality of solution phase NMR [23,24,25,26], and make it truly available as an analytical tool for solid phase synthesis. The technique is now sensitive enough that it has been used for the study of the asymmetric dihydroxylation of polymer bound olefin [27]. Due to the high quality of the spectra available now, the authors could determine directly the enantiomeric excess directly on the resin. These enantiomeric excesses were in good agreement (<1%) with whose determined by more time consuming procedure: cleavage, derivatisation with the carbonyldiimidazole, and analysis by chiral HPLC. The assignment of almost all of the resonances was also ascertained by the combination of several 2D NMR experiments.
NMR has a number of advantages over IR. It does not require undisturbed spectral regions or suitable functional groups, and is truly a non-destructive method. However, it is time consuming and special probes are needed. The weakness of signal has been circumvented by use of expensive enriched 13C tagging groups.
Mass spectrometry:
Recent reviews have been published on the application of mass spectrometry in combinatorial chemistry [28,29]. Our group has demonstrated the efficiency of the direct analysis on the resin by matrix-assisted laser desorption/ionisation time of flight techniques (MALDI TOFMS) [30,31], with a diverse array of linkers [32].
Other Methods:
Gravimetric analysis is based on the weight gain of the resin after reaction. However, the weight gain in each step of the synthesis is too small compared to the resin backbone to be accurate. Moreover, some solvent and impurities can be trapped into the resin.
Electron Spin Resonance (ESR): Katritzki et al. have reported the special preparation of spin labelled styrene divinylbenzene copolymer to quantify accurately resin loading [33]. ESR has also been used to show the mobility of bound substrate is dependent on the extent of swelling [34].
Combustion elemental analysis can suffer from the same drawbacks as gravimetric analysis, but Yan et al. have made a systematic investigation on the role of combustion elemental analysis in the quantitative monitoring solid phase synthesis. First they applied the method to the direct analysis of 9 common resins and 8 resin bound compounds [35]. The study showed that on all the samples analysed, the method was remarkably reproducible (variation <3%) and the relative error from the expected value was under 5% for most of the samples, which compares well with other established photometric method (Kaiser’test [14]), release of HOBt, dye coupling method [36,37]).
- II) Applications of Infrared Methods to “on solid phase analysis”
Yan et al. have contributed to the development of IR techniques, especially single bead microspectroscopy and ATR, applied to solid phase synthesis [9, 11].
Many IR methods have been developed by different groups, to monitor or analyse reactions on the solid support. IR spectroscopy was first used in a qualitative way. Monitoring either the appearance or disappearance of an IR chromophore in a molecule can provide a straightforward method for following a solid phase reaction. Disappearance is easier to monitor since intensity of an absorption band is difficult to quantify.
A single bead extracted randomly from the batch is to be considered as representative of the entire population as demonstrated by Yan et al. on a systematic study [38].
Some difficulties lie in the weakness of the IR signals of the compound of interest compare to the strong background signal due to the resin backbone. IR signals to be reliable must lie outside these regions. For example functions such as C-D stretching (2300-2200 cm-1), imide (1730 cm-1), nitro groups (1360 cm-1), azide (2108 cm-1), cyanide (2225 cm-1) are all clearly visible.
The KBr method:
The KBr pellet method was naturally first applied to the analysis of resin bound compounds. Indeed, the compound on the resin is in a solid state and a simple IR apparatus recording the transmitance is needed [8,39,40] . Nowadays, despite improved techniques, the KBr method is still widely used [41,42,43,44,45] since it provides a rapid qualitative answer to follow derivatisation of the resin [44] or the progression of a reaction [42, 45].
Recently, Jung et al have realised a map [41] of a library synthesised by the “split and mix” strategy. For this, they took advantage of the immobilisation of the beads in the crystalline matrix. By choosing particular IR chromophores and scanning at their absorption wavelength, they were able to assign the structure of all resin-bound compounds, creating a 2D map of their small library.
However, the KBr method has some limitations. The quality of the spectra depends on resin: better spectra are obtained with polystyrene resin rather than with the TentaGel matrix [43]. In some cases, the size of particles (generally 80µm instead of 0.5µm ideally) can cause light scattering. The preparation of the sample for analysis consumes several milligrams of resin, which can disturb the repartition of compound in the library. Finally, the presence of water in the KBr disc restricts the spectral region. Loss of informations for acids, alcohol and amides is common. When indications about these functions are needed DRIFT-IR is used [46].
The DRIFT and PAS methods:
DRIFT (Diffuse Reflectance Infrared Fourrier Transform Spectroscopy) [47] and PAS(Photoacoustic spectroscopy)[48]. DRIFT is a good method together with IR microspectroscopy to assess the presence of hydroxyl groups on resin. A comparison between IR techniques has been published earlier in this journal [49].
- a) DRIFT is based on the analysis of diffuse reflectance radiation. When infrared radiation is flashed onto a solid surface, depending on the characteristics and environment of the surface, this incident ray may be absorbed, specularly reflected (without modification, as on a mirror), internally reflected or diffusely scattered over a wide area. The detector is specially designed to maximise the collection of this latter energy, while minimising the other components (Figure 3)
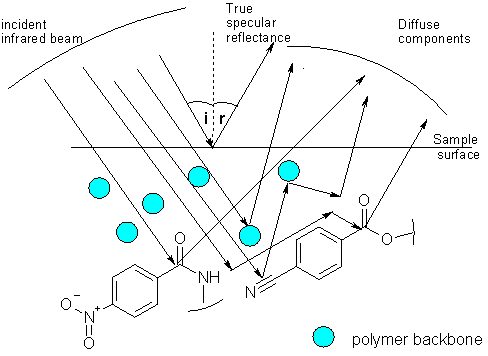
Figure 3. Schematic of some process leading to diffuse reflectance in a solid
DRIFT enables IR spectra to be recorded on diffusely scattering solids without suffering from the difficulty of sample preparation. It allows the direct observation of chemical modification on the surface and therefore is particularly suitable for monitoring reactions on pins or crowns [50]. DRIFT was used to analyse polymer films grown on platinum electrodes to prove that functional groups such as esters or nitro groups were not damaged during the oxidative polymerisation [51]. It has proved to be useful to follow derivatisation of polyester polymers (trans esterifications and amidations).
Technically, beads are placed in a metallic couple at the focal point of the diffuse reflectance accessory, under an inert atmosphere. This analysis can be automated, and has been applied to observe the disappearance of an azido stretch (2108 cm-1) in the synthesis of azasugars [52]. This method (and functionality) were also used to monitor the synthesis of a library of aspartic acid protease inhibitors [53].
- b) PAS is a non-destructive method to analyse a gas, liquid or solid. It has been mainly used to study inorganic surfaces, but its applicability to solid phase has been demonstrated [45]. The principle of PAS relies on heat transfer. The modulated infrared radiation is directed towards the sample. Absorbed radiation is transformed into heat (analogous to the green house effect). This thermal wave is then transmitted to the surrounding inert gas resulting in local pressure variations, which are detected by a sensitive microphone. The technique is less sensitive to interference such as light scattering or reflection since it does not record the resulting infrared wave, but is dependent on thermal diffusion. The main inconvenience of both methods is that the analysis chamber must be flushed with an inert gas to avoid interferences with atmospheric pollutants. Although it is really a non-destructive method, a large amount of resin is needed. A DRIFT micro version has been developed, using a smaller sample compartment. Despite a lower resolution, no significant loss of information compared to normal DRIFT was demonstrated [54].
FTIR microspectroscopy:
FTIR microspectroscopy is the first IR method developed for single bead analysis. It requires an IR microscope equiped with a very sensitive detector (this kind of apparatus is quite specialised and not widely available). A major improvement in the resolution of the IR spectra has been noticed by Yan et al. when the bead is flattened, especially on Merrifield based resins [55]. It is the most sensitive IR method and high-resolution spectra are obtained. Yan et al. were able to study the hydrogen bonding resulting from site-site interactions of hydroxyl groups in the resin. This factor is of special importance since the ability to insulate molecules from each other by attaching them to an inert and rigid matrix would have been a good alternative to the high dilution effect. Nevertheless, it has been shown that site-site interactions occur even in highly cross-linked (rigid) resins [39].
Usually, these studies are difficult to achieve because the absorption band of the hydroxyl group is very broad. However, different IR spectras, taken from single flattened bead, for Wang resin, tritylhydroxyl resin and ethylene glycol trityl resin, were recorded and showed two bands: a sharp one at 3580 cm-1 and a broad one at 3420 cm-1. They were attributed respectively to the vibration of the free hydroxyl group and to the hydrogen bonded hydroxyl group. The difference of intensity between these two bands was in accordance with the fact that a bulky surrounding (such as the trityl group) favoured the non-bonded hydroxyl [56].
When site-site interactions occur, using a large excess of reagent with respect to the number of functional groups on the bead is crucial to minimise cross coupling. Using a diacid chloride for the esterification of an alcohol onto resin it was found that 10-fold excess suppressed the cross-linking observed when only two equivalents are used. The effect of the diacid length being marginal, it is thought that site-site interactions occur because of the dynamic structure of the polymer backbone (when it is more rigid less interactions occur)[39].
The same conclusion was reached by studying site availability. In an oxidation experiment, as the reaction progresses, the population of non bonded hydroxyl groups increases since the previously bonded hydroxyl groups became free as some of its neighbours were oxidised to aldehyde [56,57].
Using the same system, Yan and Li also studied for the first time quantitatively seven reaction kinetics on the common polystyrene and TentaGel resins. Their interesting results contradict the popular presumption that TentaGel resin always allows faster reactions compared to polystyrene support. For example, the oxidation of an alcohol to an aldehyde is faster on TentaGel, but opening of oxazolidinones is 18 time faster on polystyrene resin than on TentaGel [58,59] .
Esterifications on Merrifield and TentaGel resin also gave interesting results [60]. The SN2 reaction was monitored by removing a drop of the suspension solution every 30 min., and after washing, an IR spectrum of the flattened beads recorded. The determination of reaction rates has shown that the reaction was faster on Merrifield resin than on the TentaGel and even more rapid than the one in solution. This could be attributed to the high local concentration of reagent in the polystyrene bead.
In the same publication a comparison of kinetics between the surface and the interior of the bead was also undertaken using a combination of IR flattened bead in the transmission mode and the ATR method (Attenuated Total reflection) or Internal Reflection Spectroscopy.
ATR method (Attenuated Total reflection) or Internal Reflection Spectroscopy
This technique uses the fact that the beam penetrates only 2 µm, before reflection and collection of the beam. The principle and some applications have already been reported in this journal [61]. As it is a surface sensitive technique, quality of ATR spectrum is dependent on sample contact rather than sample thickness. When applied to polymer bound-molecules, the detection limits has been estimated to 130 femtomoles [60]. ATR has proved to be useful for other applications. As it was normally assumed that bead chemistry was sensitive to diffusion control, a more rapid reaction should occur on the surface bead. The transmission mode will indicate the progression on the whole bead, whereas the ATR objective will furnish indication from the reaction taking place on the surface. Three studies (2 esterifications and 1 nucleophilic substitution) were compared using both methods, and the results demonstrate that the reactions proceed at a comparable rate [60].
Jung et al. used FT IR ATR spectroscopy to study the kinetics of cyclo addition between a polymer bound nitrone and an olefin in solution, following the imide absorption (1715 cm-1) normalised to the amide absorption of the linker (1682 cm-1) [62].
By its specificity, this method is particularly useful to study the reaction taking place on planar support. For example, the Fmoc deprotection of rink amide linker and the synthesis of a small peptide on a crown were monitored in this way [47, 50].
Examples from the laboratory
In our laboratory researchers routinely use the ‘Golden Gate diamond ATR accessory’ [For a description see IJVS (www.irdg.org/ijvs) 2, 2, 3 ]to check, or follow transformation on the resin. For example, the following figure represents the IR spectra of a diester derived polystyrene resin (green spectra). The second spectra (purple) show the result of reduction of the ester. As the ATR is however less sensitive for alcohol (as already demonstrated) the main clue is to follow the disappearance of the ester absorption at 1731 cm-1 (Figure 4).
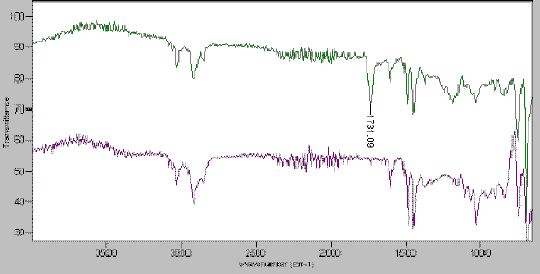
Figure 4. Reduction of an ester
Another example shows the transformation of an ester bond into an amide bond during the solid phase synthesis of dendrimers. The absorption band at 1735 cm-1disappears as the ester is treated with a diamine. The amide is characterised by the appearance of the strong absorption band at 1663 cm-1 and the broad band at 3297 cm-1 attributed to the stretching of the N-H bond of both the free amine and the primary amide (Figure 5).
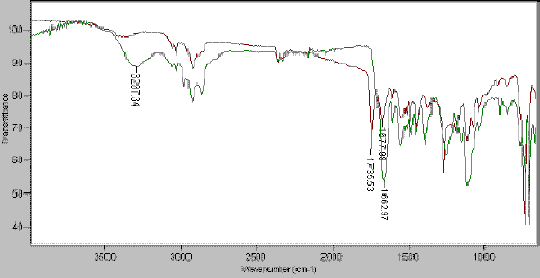
Figure 5.Formation of an amide
RAMAN Spectroscopy
This method is less used for routine structure determination than Infrared spectroscopy but is often used for the detection of certain functional groups as a complement to IR, if the functional groups are well chosen. Raman spectroscopy is particularly advantageous to the combinatorial chemist for analysis of a mixture. This time, the sample is irradiated with monochromic IR beam, and the scattered beam (composed from the parent component and the attenuated one) is then examined. The difference of frequency between the parent line and the Raman line is the frequency of the corresponding vibration. Faurskov Nielsen et al. have used Near Infrared Raman spectroscopy to follow the solid phase peptide synthesis on TentaGel of a decapeptide using the Fmoc strategy. By the observation of the spectral change in the region of the amide, the authors were able to follow the formation of b-sheets, as the peptide was growing (after 6 residues). The occurrence of this b-sheet was also responsible for the partial removal of the Fmoc protecting group upon standard treatment with piperidine in DMF [63].
By choosing carefully IR and Raman tags (nitrile, phenol, alkyne) Rahman et al. were able to unambiguously identify the different template chosen randomly by a combination of IR and Raman microspectrometry [64]. This strategy seems promising since it can be used for code reading directly from the resin. However, the strategy is still limited to a small number of molecules and increasing the number of coding elements means using a combination of other analytical methods to read the code (see also [41]).
Conclusion
Infrared spectroscopy proves to be a method of choice for quickly monitoring reactions occurring in the solid phase, as the method demonstrates several advantages over it’s competitors:
a) IR spectrometers are available in all organic laboratories.
b) The method is usable by all chemists, not only experienced analytical chemists.
c) It is a rapid method since it is an “on bead analysis” and it does not require sample preparation (except for KBr pellets), so a fast feedback for reaction optimisation becomes possible.
Among the techniques presented, ATR is a bit less sensitive than DRIFT or microspectroscopy for the detection of hydrogen bonding functions, but is much cheaper and based on good quality spectra. The results are very rapidly acquired (spectra can be recorded in less than one minute) and easy to get from a diamond apparatus. However, chemists will be unable to monitor a reaction on solid phase with IR spectroscopies if no change of spectral absorption in a functional group occurs. Thus, IR will be best exploited when used in combination with other methods (NMR, mass spectrometry, HPLC…).
References
- Merrifield R. B., (1963): Solid phase peptide synthesis. I. The synthesis of a tetrapeptide, J. Am. Chem. Soc., 85, 2149-2154
- a) Fruchtel J. S., Jung G. (1996): Organic chemistry on solid support, Angew. Chem. Int. Ed. Eng., 35, 17-42
b) Balkenholl F., Von dem Bussche-Hunnefeld C., Lansky A., Zechel C. (1996): Combinatorial Synthesis of small molecules, Angew. Chem. Int. Ed. Eng., 35, 2288-2337
c) Special issue of Acc. Chem. Res., 1996, 29, 3, 112-170 - a)Koshla C. (1998):Combinatorial biosynthesis: New tools for the medicinal chemist, Chemtracts-Organic chemistry, 11, 1-15
b) Liu D. R., Schultz P. G. (1999): Generating new molecular function: A lesson from nature, Angew. Chem. Int. Ed. Eng., 38, 36-54 - a) Gallop M. A., Barrett R. W., Dower W. J., Fodor S. P. A., Gordon E. M. (1994): Application of combinatorial technologies to Drug discovery. 1. Background and peptide combinatorial libraries, J. Med. Chem., 37, 9, 1233-1251
b) Gordon E. M., Barrett R. W., Dower W. J., Fodor S. P. A., Gallop M. A. (1994): Application of combinatorial technologies to Drug discovery. 2. Combinatorial organic synthesis, library screening strategies, and future directions, J. Med. Chem., 37, 9, 1386-1401.
c) Terrett N. K., Gardner M., Gordon D. W., Kobylecki R. J., Steele J. (1995): Combinatorial synthesis – The design of compound libraries and their application to drug discovery, Tetrahedron, 51, 30, 8135-8173 - Furka A., Sebestyen F., Asgedom M., Dibo G. (1991): General method for rapid synthesis of multicomponents peptide mixtures, Int. J. Protein Res., 37, 487-493
- Lam K. S., Lebl M., Krchnak V. (1997): The “One-bead-one-compound” combinatorial library method, Chem. Rev., 97, 411-448.
- Czarnik A. W. (1998): Solid phase supports are like solvents, Biotechnol. Bioeng. (Comb. Chem.), 61, 77-79
- Frechet J. M., Schuerch C. (1971): solid phase synthesis of oligosaccharides. I. Preparation of the solid support. Poly[p-(1-propen-3-ol-1-yl)styrene], J. Am. Chem. Soc., 93, 2, 492-496
- Yan B. (1998): monitoring the progress and the yield of solid phase organic reaction directly on resin support, Acc. Chem. Res., 31, 621-630
- Gallop M. A., Fitch W. L. (1997): New methods for analysing compounds on polymeric supports, Curr. Opinion in Chem. Bio., 1,1 94-100
- Yan B., Gremlich H-U., Moss S., Coppola G. M., Sun Q., Liu L. (1999): A comparison of various FTIR and FT Raman methods: application in the reaction optimisation stage of combinatorial chemistry, J. Comb. Chem., 1, 46-54
- Egner B. J., Bradley M. (1997): Analytical techniques for solid phase organic synthesis and combinatorial synthesis, Drug Discovery Today, 2, 102-110
- Swali V., Bradley M. (1997): Analytical techniques for single bead analysis in combinatorial chemistry, Anal. Comm., 34, 15H-18H
- Sarin V. K., Kent S. B. H., Tam J. P., Merrifield R. B. (1981): Quantitative monitoring of solid phase peptide synthesis by the ninhydrin reaction, Anal. Biochem., 117, 147-157
- Wells N. J., Davies M., Bradley M. (1998): cleavage and analysis of material from single resin beads, J. Org. Chem., 63, 643-6431
- Krchnark V., Vagner J., Safar P., Lebl M. (1988): Non-invasive continuous monitoring of solid phase peptide synthesis by acid-base indicator, Coll. Czech. Chem. Comm., 53, 2542-2548.
- Virgilio A. A., Ellman J. A. (1994): Simultaneous solid phase synthesis of ?-turn mimetics incorporating side-chain functionality, J. Am. Chem. Soc., 116, 11580-11581
- Blossey E. C., Cannon R. G., ford W. T. , Periyasamy M., Mohanraj S. (1990) : synthesis reactions, and 13C FT NMR spectroscopy of polymer-bound steroids, J. Org. Chem., 55, 4664-4668
- Look G. C., Holmes C. P., Chinn J. P., Gallop M. A. (1994): Method for combinatorial organic synthesis: the use of fast 13C NMR analysis for gel phase reaction monitoring, J. Org. Chem. 59, 7588-7590
- Kanemitsu T., Kanie O., Wong C-H. (1998): quantitative monitoring of solid phase synthesis using gated decoupling 13C NMR spectroscopy with a 13C-enriched group and an internal standard in the synthesis of sialyl Lewis tetrasaccharide, Angew. Chem. Int. Ed. Eng., 37, 24, 3415-3418
- Anderson R. C., Jarema M. A., Shapiro M. J., Stokes J. P., Ziliox M. (1995): Analytical technique in combinatorial chemistry: MAS CH correlation in solvent swollen resin, J. Org. Chem., 60, 2650-2651
- Fitch W. L., Detre G., Shoolery J. N., Keifer P. A. (1994): High resolution 1H NMR in solid phase organic synthesis, J. Org. Chem., 59, 7995
- Anderson R. C., Stokes J. P., Shapiro M. J. (1995): Structure determination in combinatorial chemistry : utilisation of Magic Angle Spinning HMQC and TOCSY NMR spectra in the structure determination of Wang-bound Lysine, Tet. Lett., 36, 30, 5311-5314
- Pursch M., Schlotterbeck G., Tseng L-H., Albert K., Rapp W. (1996): Monitoring the reaction progress in combinatorial chemistry: 1H MAS NMR investigations on single macro beads in the suspended state, Angew. Chem. Int. Ed. Eng., 35, 23-24, 2867-2869
- Chin J., Fell B., Pochapsky S., Shapiro M. J., Wareing J. R. (1998): 2D SECSY NMR for combinatorial chemistry. High resolution MAS Spectra for resin-bound molecules, J. Org. Chem., 63, 1309-1311
- Ruhland T., Andersen K., Pedersen H. (1998): Selenium-linking strategy for traceless solid phase synthesis: direct lading , aliphatic C-H bond formation upon cleavage and reaction monitoring by gradient MAS NMR spectroscopy, J. Org. Chem., 63, 9204-9211
- Riedl R., Tappe R., Berkessel A. (1998): Probing the scope of the asymmetric dihydroxylation of polymer-bound olefins. Monitoring by HRMAS NMR allows for reaction control and on-bead measurement of enantiomeric excess, J. Am. Chem. Soc., 120, 8994-9000
- Siuzdak G., Lewis J. K. (1998): Application of mass spectroscopy in combinatorial chemistry, Biotechnology and bioengineering (combinatorial chemistry), 61, 2, 127-134
- Swali V., Langley G. J., Bradley M. (1999): Mass spectrometric Analysis in combinatorial chemistry, Current Opinion in Chemical Biology, in press.
- Egner B. J., Langley J. G., Bradley M. (1995): SPC: Direct monitoring by matrix–assisted laser desorption/ionisation time of flight mass spectrometry. A tool for combinatorial chemistry, J. Org. Chem., 60, 2652-2653.
- Egner B. J., Bradley M. (1997): Monitoring the solid phase synthesis of analogues of Lysobactin and katanosins using in situ MALDI-TOF-MS, Tetrahedron, 53, 41, 14021-14030
- Egner B. J., Cardno M., Bradley M. (1995): Linkers for combinatorial chemistry and reaction analysis using solid phase in situ mass spectrometry, J. Chem. Soc., chem. Commun., 2163-2164
- Katritzki A. R., Belayakov S. A., Strah S., Cage B., Dalal N. S. (1999): Preparation of spin labelled styrene divinylbenzene copolymers and a new approach for the quantitative determination of the resin loading using ESR spectroscopy, Tet. Lett., 40, 407-410
- Regen S. L. (1975), Macromolecules, 8, 689
- Yan B., Jewell C. F., Meyers S. W. (1998): Quantitatively monitoring of solid phase organic synthesis by combustion elemental analysis, Tetrahedron, 54, 11755-11766.
- Yan B., Li W. (1997): Rapid fluorescence determination of the absolute amount of aldehyde and ketone groups on resins supports, J. Org. Chem., 62, 9354-9357.
- Li W., Yan B. (1997): A direct comparison of the mixing efficiency in solid phase organic synthesis by single bead IR and fluorescence spectroscopy, Tet. Lett., 38, 37, 6485-6488
- Yan B., Kumaravel G., Anjaria H., Wu A., Petter R. C., Jewell C. F. JR., Wareing J. R. (1995): Infrared spectrum of a single resin bead for real-time monitoring of solid phase reaction, J. Org. Chem., 60, 5736-5738
- Scott L. t., Rebek J., Ovsyanko L., Sims C. L. (1977): Organic chemistry on the solid phase. Site-site Interactions on functionalized polystyrene, J. Am. Chem. Soc., 99, 2, 625-626
- Farrall M. J., Frechet M. J. (1976): Bromination and lithiation: two important steps in the functionalization of polystyrene resins, J. Org. Chem., 41, 24, 3877-3882
- Haap W., Walk T. B., Jung G. (1998): FTIR mapping-A new tool for spatially resolved characterisation of polymer-bound combinatorial compound libraries with IR microscopy, Angew. Chem. Int. Ed., 37, 23, 3311-3314
- Gordeev M. F., Patel D. V., Gordon E. M. (1996): Approaches to combinatorial synthesis of heterocycles: a solid phase synthesis of 1,4-dihydropyridines, J. Org. Chem., 61, 924-928
- Hauske J. R., Dorff P. (1995): A solid phase Cbz chloride equivalent–a new matrix specific linker, Tet. Lett., 36, 10, 1589-1592
- Stranix B. R., Ping Gao J., Barghi R., Salha J., Darling G. D. (1997): Functional polymers from (vinyl)polystyrene. Short routes to binding functional groups to polystyrene resin through a dimethylene spacer: bromine, sulphur, phosphorus, silicon, hydrogen, boron and oxygen, J. Org. Chem., 62, 8987-8993
- Russell K., Cole D. C., McLaren F. M., Pivonka D. E. (1996): Analytical techniques for combinatorial chemistry: qualitative infrared spectroscopic measurement of deuterium labelled protecting group, J. Am. Chem. Soc., 118, 7941-7945
- Deben I.E.A., Van Wijk T.H.M., Van Doornum E. M., Hermkens P. H. H., Kellenbach E.R. (1997): Characterisation of reaction products on solid support by FT-IR. Mikrochim. Acta, 14, 245-246
- Gremlich H-U (1998): Infrared and Raman spectroscopy in combinatorial chemistry. American Laboratory 33-39
- Gosselin F., Di Renzo M., Ellis T. H., Lubell W. D. (1996): Photoacoustic FTIR spectroscopy, a non destructive method for sensitive analysis of solid phase organic chemistry, J. Org. Chem., 61, 7880-7981
- Alexander R. (1999): FT-IR analysis of combinatorial beads and crowns http://www.irdg.org/ijvs.volume2/edition4/section1.htm
- Gremlich H-U., Berets S. L. (1996): Use of FTIR internal reflection spectroscopy in combinatorial chemistry, Appl. Spect., 50, 4, 532-536
- Passos M. S., Queiros M. A., Le Gall T., ibrahim S. K., Pickett C. J.(1997): Solid phase chemistry of electropolymers, J. Electroanalitycal Chemistry, 435, 189-203
- Chan T. Y., Chen R., Sofia M. J., Smith B. C., Glennon D. (1997): High throughput on-bead monitoring of solid phase reaction by Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS), Tet. Lett., 38, 16, 2821-2824
- Kick E.K., Ellman J. A. (1995): Expedient method for the solid phase synthesis of aspartic acid protease inhibitors directed toward the generation of libraries, J. Med. Chem., 38, 1427-1430
- Deben I., Goorden J., Van doornum E., Ovaa H., Kellenbach E. (1998): MicroDRIFT: rapid and efficient IR analysis of SPOCR, Eur. J. Org. Chem., 63, 697-700
- Yan B., Kumaravel G. (1996): Probing solid phase reaction by monitoring the IR bands of compounds on a single “Flattened” resin bead, Tetrahedron, 52, 3, 843-848
- Yan B., Sun Q. (1998): Crucial factors regulating site interactions in resin supports determined by single bead IR, J. Org. Chem., 63, 55-58
- Yan B., Sun Q., Kumaravel G., Jewell C. F. JR., Wareing J. R (1996): Real-time monitoring of the catalytic oxidation of alcohol to aldehyde and ketoses on resin support by single-bead Fourier transform infrared microspectroscopy, J. Org. Chem., 61, 8765-8770
- Li W., Yan B. (1998): Effect of polymer support on kinetics of solid phase organic reaction: a comparison of polystyrene- and TentaGel- based resins, J. Org. Chem., 63, 4092-4097.
- Marti R. E., Yan B., Jarosinsky M. A. (1997): solid phase synthesis via 5-oxazolidinones. Ring-opening reactions with amines and reaction monitoring by single bead FTIR microspectroscopy, J. Org. Chem., 62, 5615-5618.
- Yan B., Fell J. B., Kumaravel G. (1996): Progression of organic reactions on solid supports monitored by single bead FTIR microspectroscopy, J. Org. Chem., 61, 7467-7472
- Combs D. (1998): The use of diamond as an ATR material, http://www.irdg.org/ijvs/volume2/edition2/section1.htm
- Haap W. J., Kaiser D., Walk T. B., Jung G. (1998): solid phase synthesis of diverse isoxazolidines via 1,3-dipolar cycloaddition, Tetrahedron, 54, 3705-3724.
- Ryttersguard J., Due Larsen B., Holm A., Christensen D. H., Faurskov Nielsen O. (1997): Monitoring of a solid phase peptide synthesis by NIR-FT-Raman spectroscopy, Spectrachimica Acta part A-molecular and Biomolecular spectroscopy, 53, 1, 91-98
- Rahman S. S., Busby D. J., Lee D. C. (1998): Infrared and Raman spectra of a single resin bead for analysis of solid phase reaction and use in encoding combinatorial libraries, J. Org. Chem., 63, 6196