A technological approach of quantitative analysis using Vibrational Spectroscopy in the polymer industry
![]()
5. A Technological Approach to Quantitative Analysis using Vibrational Spectroscopy in the Polymer Industry
Sharon Cooke, Catherine Deeley and Mark Billingham
Perkin-Elmer Limited,
Post Office Lane,
Beaconsfield, Bucks.
HP9 1QA
United Kingdom
Telephone: +44 (0) 1494 676161
Mid infrared spectroscopy has a long history in polymer science. At the fundamental level, the spectra are well understood whilst those of unknowns are readily identified using `fingerprinting’. Vibration spectra of polymers are sensitive to polymer morphology (crystallinity and/or orientation) and such information can be obtained using mid infrared spectroscopy. Quantitative analysis is also possible using mid infrared. Several standard procedures are available for these analyses but they are often time consuming and expensive because they invariably require, as part of the analysis, the production of reproducible film samples. In fact, one of the main problems when using mid infrared spectroscopy for polymer analysis has been the difficult and time consuming sample preparation. Traditionally, the standard procedure for MIR transmission sampling is to produce a film ~ 30 mm thick either by microtoming or by the use of a hot press and a film making accessory. Unfortunately, microtoming is highly skilled, time consuming and expensive whereas melting and film making can frequently ruin the sample if, for example, the purpose of the analysis is to measure orientation or crystallinity.
Raman spectroscopy is an alternative method of investigating the vibrational behaviour of polymers but is far less used than mid infrared spectroscopy. The reasons for this are largely historical but with the development of Fourier Transform (FT) Raman spectroscopy spectra can be recorded very rapidly directly on samples with little or no sample preparation. Samples as different as a fibre, a lump or even a screw driver handle can be studied almost instantly. Unfortunately, the main weakness of Raman spectroscopy is its poor repeatability in quantitative analysis and perhaps it is this that restricts its acceptability to industrial analysts. This is unfortunate because the spectra are of very high quality, constitute an excellent fingerprint and are often well understood in the literature. Morphological features of a specimen are clearly indicated in the Raman spectrum as they are in the mid infrared.
In contrast, near infrared spectra are very different from those obtained using mid infrared and Raman methods. The spectra arise from overtones of the fundamental frequencies and so the spectra consist of many overlapping bands. This results in very complex spectra which tend to be unresolved and almost featureless and to the eye, often constitute no useful fingerprint. However, it must be accepted that in the quantitative measurement of complex mixtures (for example in the food or agricultural industries or more recently in the pharmaceutical field) near infrared spectroscopy has proved to be a valuable tool and its application is expanding very rapidly. The NIR analysis of such complex spectra typically relies on a variable which follows the property change of interest and simply relates the two in some simple manner, say by a linear or simple quadratic relationship. The advantage of NIR spectroscopy over other analytical techniques is cost per sample – sample preparation is often non existent and the complete analysis can be performed very rapidly. The data analysis is mathematically sophisticated but the analysis is routine, reliable, rapid and if carefully designed, almost fool-proof. Near infrared spectroscopy is normally carried out in transmission or reflection. Solid samples are presented as films, lumps, sheets or powders in their original containers with little or no sample preparation. Mid infrared and Raman spectroscopy have been seen, perhaps unfairly, to lag behind near infrared with respect to sample preparation and ease of analysis but, in appropriate experimental circumstances, these techniques can rival near infrared in speed and convenience. The aim of this article is to demonstrate how the different technological approaches mentioned above can be applied to quantitative analysis in the polymer industry.
Sampling alternatives for mid infrared spectroscopy do exist; one can make reflection measurements either specularly or by diffuse reflection. In both, the surface condition and presence of fillers are highly significant and hence tend not to be reproducible. Also, the spectra recorded may need mathematical processing to make them recognisable. Attenuated Total Reflectance (ATR) is a possibility but is initially unattractive because polymers are often hard and rigid and hence it is difficult to produce the essential optical contact between the sample and the ATR crystal. Recently, this problem has been reduced thanks to the development of single reflection diamond ATR accessories. Hence, hard or soft specimens are squeezed against one surface of the diamond prism (~1mm sq.). These accessories are very easy to use and sampling takes only seconds.
Samples of ethylene and vinyl acetate copolymers with a wide range of concentrations, were supplied by ICI Wilton Research Centre in an attempt to analyse the vinyl acetate content by mid infrared spectroscopy. Initially the mid infrared spectra were recorded with no sample preparation. All spectra were recorded on a Perkin Elmer Spectrum GX fitted with the Golden Gate diamond ATR accessory. Data was collected at 4 cm-1 resolution over a period of one minute scanning time. Each polymer was squeezed onto the diamond ATR to give enough pressure to ensure good spectra were obtained. Spectra of the EVA calibration samples are illustrated in Figure 1 where the different levels of vinyl acetate can clearly be seen. The spectra obtained were mathematically analysed using a partial least squares algorithm using Spectrum Quant+ software. Further EVA samples were supplied by the University of Southampton to use as an independent validation set. The data was referenced by NMR spectroscopy. Following the PLS analysis a standard error of prediction of 0.5 was obtained using 2 PLS factors and X number of standards in the calibration. It is believed that the SEP value would improve with the addition of more standards. A regression summary is shown in Table 1 and the Estimated vs Specified plot is shown in Figure 2. The methods built were tested using independent validation samples supplied by the University of Southampton. The validation spectra were recorded and a typical prediction report is shown in Table 2. Although the predicted value of vinyl acetate is very good at 8.74% compared to the referenced value of 9%, the statistics in the report indicate that the residual ratio is quite large. This is most likely due to the difference in sample form, pellets as opposed to slabs, which were not incorporated into the calibration.
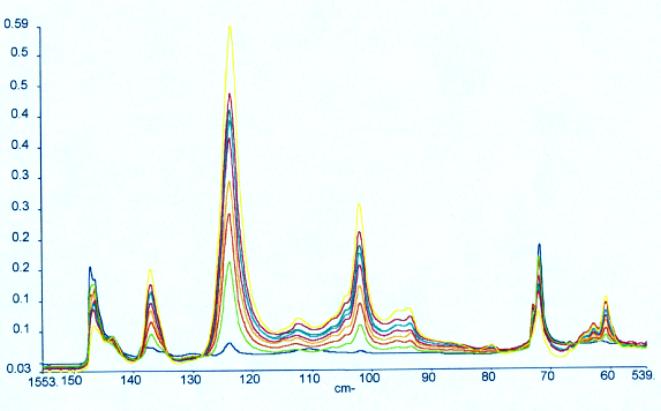
Figure 1. Mid infrared spectra of EVA polymers
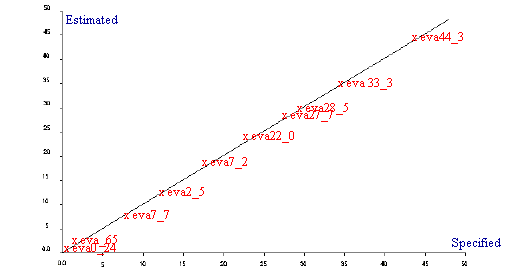
Figure 2. Estimated vs Specified plot for EVA by mid infrared
| Using 2 PLS factors for 10 samples | ||
| Std Error of Prediction | Estimate = 0.4555 | Actual = 0.499 |
| Multiple Correlation | = 0.9996 | |
| % Variance (R squared) | = 99.9265 | |
| Std Error of Estimate (SEE) | = 0.4441 | |
Table 1. Regression summary for EVA
| QUANT+ V4.10 PREDICTION RESULTS PLS1 | |||||
| Sample | PEVA_9 | ||||
| Calc. Name | R01PEA_9.SP | ||||
| Normalization | None | ||||
| Method | PEVA.MD Ver:9 ID:3284 | ||||
| RMS Error | 0.003332 A | ||||
| Peak to Peak Error | 0.0345 A | ||||
| Total M-Distance | 0.0192 | ||||
| Residual Ratio | 4.55 | ||||
| Property | Calc. Value | Deviation | R-Error | M-Distance | |
| VinAc 8.74 % 0.874 0.192 | |||||
| Prediction complete | |||||
Table 2. Prediction Report for EVA
Following the success of the EVA calibration using mid infrared spectroscopy it was decided to repeat the analysis using both Raman and NIR spectroscopy. Raman measurements were recorded on a Spectrum GX Raman and all near infrared spectra were recorded on a Perkin Elmer Spectrum IdentiCheck FT-NIR spectrometer fitted with the IdentiCheck Reflectance accessory (ICRA). Raman measurements were recorded at 4 cm-1 resolution whilst the NIR spectra were recorded at 8cm-1 resolution. The regression summaries (Table 3 and Table 4) and the Estimated vs Specified plots for both the Raman and NIR analyses are shown in Figure 3 and Figure 4. Although both techniques produce very good calibrations and would be ideal to use for the analysis of ethylene vinyl acetate polymers it can be seen that mid infrared produced the most superior results based on the samples used.
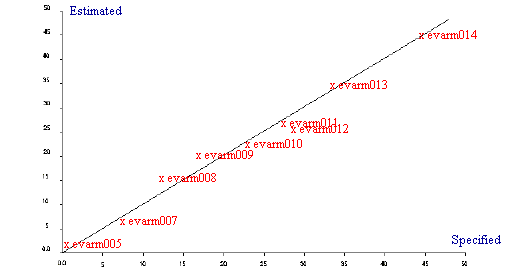
Figure 3. Estimated vs Specified plot for EVA by Raman
| 3 PLS factors for 10 samples | |
| Std Error of Prediction: | Estimate = 1.807 Actual = 2.931 |
| Multiple Correlation | = 0.9955 |
| % Variance (R squared) | = 99.0944 |
| Std Error of Estimate (SEE) | = 1.685 |
Table 3. Regression summary for EVA by Raman
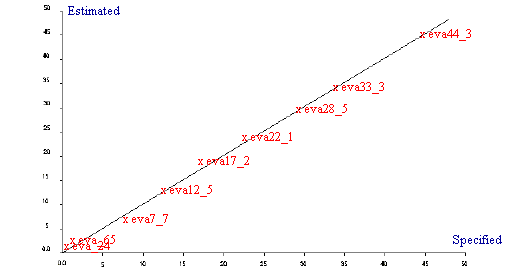
Figure 4. Estimated vs Specified plot for EVA by NIR
| 3 PLS factors for 10 samples | |
| Std Error of Prediction: | Estimate = 0.8555 Actual = 1.484 |
| Multiple Correlation | = 0.9993 |
| % Variance (R squared) | = 99.8603 |
| Std Error of Estimate (SEE) | = 0.7101 |
Table 4. Regression summary for EVA by NIR
Composition in natural rubber-gutta purcha mixtures
MIR, Raman and NIR techniques were used to analyse a second set of polymer samples supplied by the University of Southampton. The mixture of naturally occuring cis and trans polyisoprene samples that were supplied was rather unusual. Trans polyisoprene – gutta percha was historically quite important because like natural rubber, it is biologically derived, but these days is rarely encountered. The set of samples are hence laboratory oddities, but they do provide a series similar to ones encountered in the commercial world. Raman spectroscopy was the most obvious choice for this analysis because the Vc=c Raman band in unsaturated compounds is invariably strong and its frequency varies with isomer type. The main problem arises because both the cis and trans polyisoprene are tertiary olefines and hence the c=c stretching frequency in each case overlaps. It was for this reason that we decided to look at the mid infrared spectra. The spectra were recorded using as light a pressure between the sample and the diamond as possible and examples of the calibration spectra are illustrated in
Figure 5. A quantitative model was built, again using PLS. The Estimated vs Specified plot is shown in Figure 6 and the regression summary can be seen in Table 5. If excessive pressure is used the spectra can change dramatically. For example, the undiluted natural rubber specimen can crystallise. Keeping rubber for extended periods at around 0° C or stretching its crosslinked form to extension ratios beyond ~400% both induce crystallisation and this morphological change has been monitored in the past by both infrared transmission and F-T Raman methods. Figure 7 shows the crystallisation induced by the application of pressure.
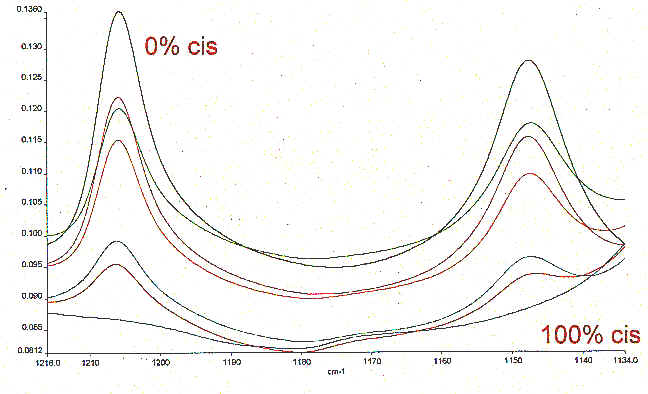
Figure 5. Calibration spectra of Natural Rubber samples by MIR
| 1 PLS factors for 6 samples | |
| Std Error of Prediction: | Estimate = 1.859 Actual = 2.284 |
| Multiple Correlation | = 0.9992 |
| % Variance (R squared) | = 99.8442 |
| Std Error of Estimate (SEE) | = 1.597 |
Table 5. Regression summary for Natural rubber by MIR
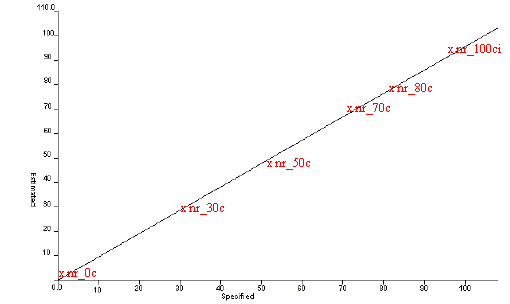
Figure 6. Estimated vs Specified plot for Natural Rubber
by mid infrared
| 2 PLS factors for 6 samples | |
| Std Error of Prediction: | Estimate = 0.8946 Actual = 1.277 |
| Multiple Correlation | = 0.9998 |
| % Variance (R squared) | = 99.9624 |
| Std Error of Estimate (SEE) | = 0.8797 |
Table 6. Regression summary for Natural rubber by NIR
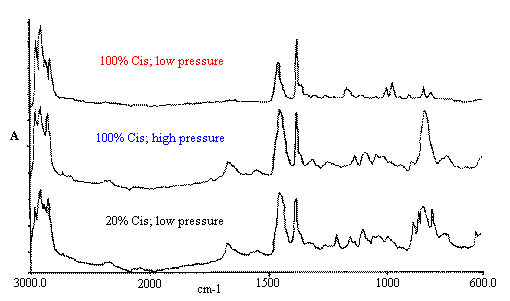
Figure 7. Natural rubber in Diamond ATR Pressure Sensitive Morphology
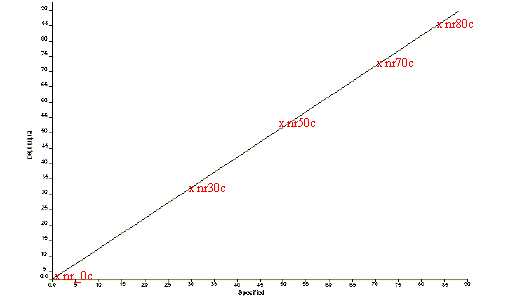
Figure 8. Estimated vs Specified plot for Natural Rubber by NIR
NIR spectra were also recorded on the same rubber samples to obtain a very good correlation between the spectra and the cis-trans ratio. The results from this analysis, shown in Figure 8 and Table 6, can be compared with those obtained using ATR (Figure 6, Table 5). Raman spectra were not recorded on the natural rubber samples.
Crystallinity in Polyether ether ketone (PEEK)
Polyether ether ether ketone is a high temperature thermoplastic which is widely used in the Aero Industry. The material can easily be made in the form of a glass and with appropriate heat treatment will crystallise up to about 50% degree of crystallinity. The polyether ketone samples supplied by ICI were used in an attempt to determine the crystallinity of such samples. The data set comprised of well referenced samples that had previously been analysed by X-Ray diffraction. Secondary measurements of crystallinity have been reported in the literature based on both mir and Raman measurements. However, a serious problem arises when Raman spectroscopy is used to determine crystallinity since degradation often takes place during heat treatment and even with near infrared laser excitation fluorescence is a problem in Raman spectroscopy. However, when these samples were scanned using mir spectroscopy with the diamond ATR accessory the problem is overcome and excellent spectra are produced. Examples of the spectra and the calibration plot are given Figure 9 and Figure 10 and a regression summary is shown in Table 7. It is clear that an excellent correlation can be generated. NIR spectra were generated but unfortunately, an acceptable calibration could not be obtained with the samples supplied and so MIR is the preferred technology in this case.
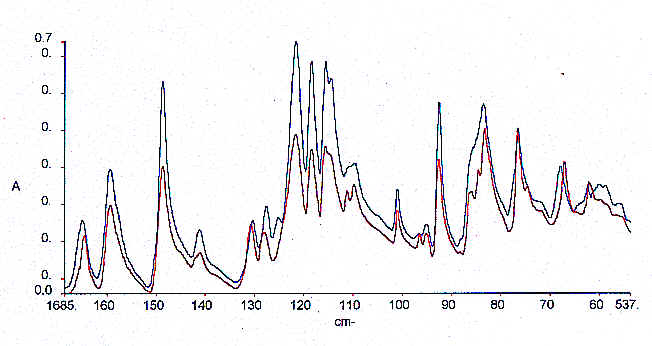
Figure 9. Extremes of calibration spectra for PEEK samples – MIR
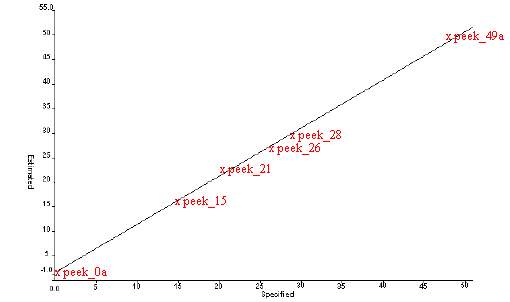
Figure 10. Estimated vs Specified plot for PEEK samples by MIR
| 2 PLS factors for 6 samples | |
| Std Error of Prediction: | Estimate = 0.5603 Actual = 0.8275 |
| Multiple Correlation | = 0.9997 |
| % Variance (R squared) | = 99.9471 |
| Std Error of Estimate (SEE) | = 0.48 |
Table 7. Regression summary for PEEK samples by MIR
Young’s Modulus in oriented polypropylene
In principle one can relate any variable to a chosen spectroscopic change from sample to sample. The variable does not have to be composition or molecular. Highly oriented polymers (materials that have been irreversibly extended) show high levels of stiffness, a property exploited in the textile industry in tyre cords and packaging. These industries need to monitor the elongation and often Young’s modulus of the materials they produce. This is done by gripping a sample, applying a load and measuring the deflection on a tensile testing machine. Gripping is problematic, the reproducibility is poor, special samples have to be made and it is normal to make several measurements and average – all very expensive and slow.
It is known that when elongated, molecular orientation occurs in a polymer the modulus increases. The correlation between orientation and modulus is complex, as is the elongation vs orientation. The degree of orientation can be measured spectroscopically but obviously the correlation between any spectroscopic effect and the modulus will be pretty obscure. Orientation in polymer films are measured spectroscopically by recording the mir spectrum in polarised light, orienting the sample with its prepared axis parallel and perpendicular to the electric vector of the radiation. Each band intensity in each spectrum is then compared and a set of dichroic ratios is generated. Since the general direction of the movement vectors of the atoms with respect to the molecular axis in at least some of the vibrational modes is known, the dichroic ratios can be used to deduce the perfection of the orientation. Although not strictly a completely relevant measurement, we show in Figure 11 the effect of aligning a sample parallel and perpendicular to the vector direction and the appearance of dichroism.
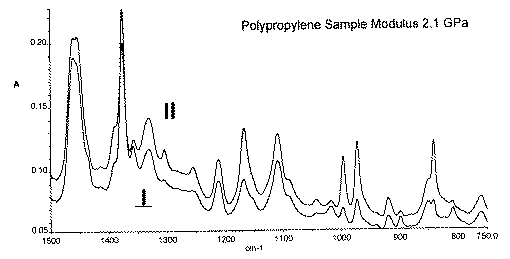
Figure 11. Effect of sample orientation in Diamond ATR
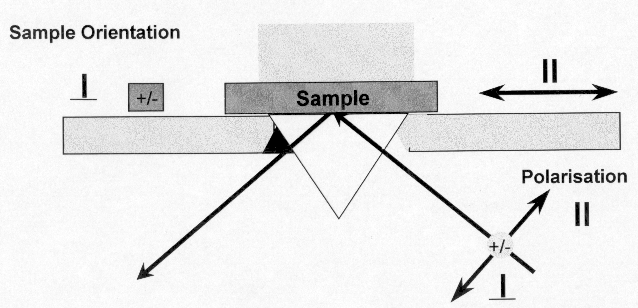
Figure 12. Diamond ATR Polarisation and sample orientation convention
An experiment was set up as in Figure 12 using the diamond ATR in polarised light. The chosen orientation is clearly identified in the caption. Samples of oriented polypropylene extended to different degrees and of known modulus were supplied by Dr. Shilin Lu of the University of Southampton. Each sample was examined with its molecular axis parallel to the front of the instrument – (perpendicular) (Figure 13) and then arranged normal to the front of the instrument – (parallel) (Figure 14). The set of spectra in each series were submitted to PLS analyses using Spectrum Quant+ and the results are reported below in Table 8a and Table 8b. Clearly the correlation between the spectral variables and Young’s Modulus is outstanding and the spectra are very simple indeed to record.
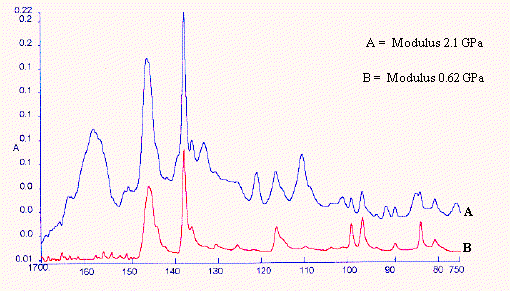
Figure 13. Polypropylene samples – perpendicular
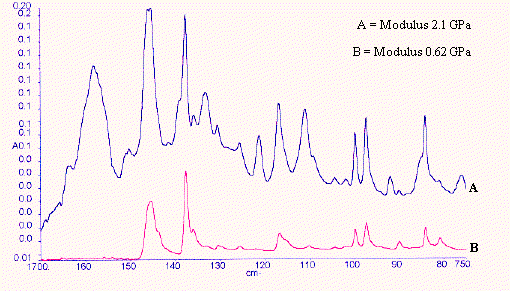
Figure 14. Polypropylene samples – parallel
| 1 PLS factors for 6 samples | |
| Std Error of Prediction: | Estimate = 0.06049 Actual = 0.07991 |
| Multiple Correlation | = 0.9928 |
| % Variance (R squared) | = 98.5565 |
| Std Error of Estimate (SEE) | = 0.05963 |
Table 8a. Tensile modulus by Diamond ATR.
Polypropylene sample Perpendicular
| 3 PLS factors for 7 samples | |
| Std Error of Prediction: | Estimate = 0.1098 |
| Multiple Correlation | = 0.9925 |
| % Variance (R squared) | = 98.5038 |
| Std Error of Estimate (SEE) | = 0.1048 |
Table 8b. Tensile modulus by Diamond ATR.
Polypropylene sample Parallel
By adopting the logic universally accepted by near infrared users, one can indeed use simple, rapid and easy-to-use sampling methods in the mid infrared or Raman for both qualitative and quantitative analysis of a wide range of polymers. Their use will significantly reduce the cost of analysis since the experimental data can be acquired from an unknown in a few minutes and the mathematical processing is automatic and almost instantaneous.
Editor’s Note: We will cover the mathematical analysis of results – so called chemometrics in a future edition.