Identification of Compounds in HPLC-Eluates by Raman Spectroscopy
![]()
7. Identification of compounds in HPLC-eluates by Raman Spectroscopy
Roland Steinert, Hans Bettermann* and Karl Kleinermanns
Institut für Physikalische Chemie und Elektrochemie Heinrich-Heine-Universität Düsseldorf, Geb. 26.43.02 D-40225 Düsseldorf, Germany
E-mail: betterma [at] c2.rz.uni-duesseldorf.de
* author to whom correspondence should be addressed
Introduction
The versatility of Raman spectroscopy in determining molecular properties has been proved by solving innumerable problems over many decades. The success of Raman spectroscopy is based on its simple instrumentation which consists of a powerful light source, a well-dispersing monochromator or interferometer and a sensitive detection device. Furthermore, no special efforts have to be made to prepare samples for Raman analysis.
In addition, modern computers enable the prediction of vibrational spectra very rapidly† so that the analysis of transitions is theoretically assisted and this gives a comprehensive interpretation of Raman spectra.
† Editor’s Note : we plan a special edition devoted to the calculation of vibrational frequencies early in Vol III.
Compared to molecular fluorescence emissions, Raman signals are notourisly weak. As a rule of thumb, one million laser photons generate only one Raman photon! The magnitude of the scattered radiation per unit solid angle depends linearly on the intensity of the excitation laser light source, the number of exposed molecules, the fourth power of the absolute scattering wavenumber, and the sum of the squared elements of the scattering tensor. The smallness of the product determines the small yield of Raman photons. The magnitude of scattering tensor elements can be increased by matching the frequency of the laser to an optically allowed transition. In this case, certain scattering signals of the molecule being investigated can be strongly enhanced by resonance effects*. Beside this property, the increase of the frequency and the power of the excitation source as well as the extension of the solid angle of the scattered light are also tools to enhance the scattering intensity.
* Another subject for a future edition.
In this article we are concerned with the best possible detectability of Raman signals by improving the instrumental components especially the imaging of the scattered light [1]. In addition, the imaging conditions of our setup enables us to record scattering signals from small-sized sample. As a result, it is possible to consider Raman spectroscopy as a detection device for HPLC [2-6].
In HPLC using normal conditions, structural isomers often have the same retention times. Thus, it can sometimes be hard to decide which isomer or indeed how many compounds generate a signal in a chromatogram. Since electronic spectra of compounds in the liquid phase have large halfwidths and show only slight differences between them, fluorescence spectroscopy and absorption spectroscopy are overtaxed as conventional detection methods in HPLC. The answer, we think is Raman spectroscopy.
Experimental Approach
The setup (Figure. 1) can be divided into five functional sub-units, which will be discussed successively: the excitation, the sample cell, the light collection unit, the light-dispersive system, and finally the light detection assembly.
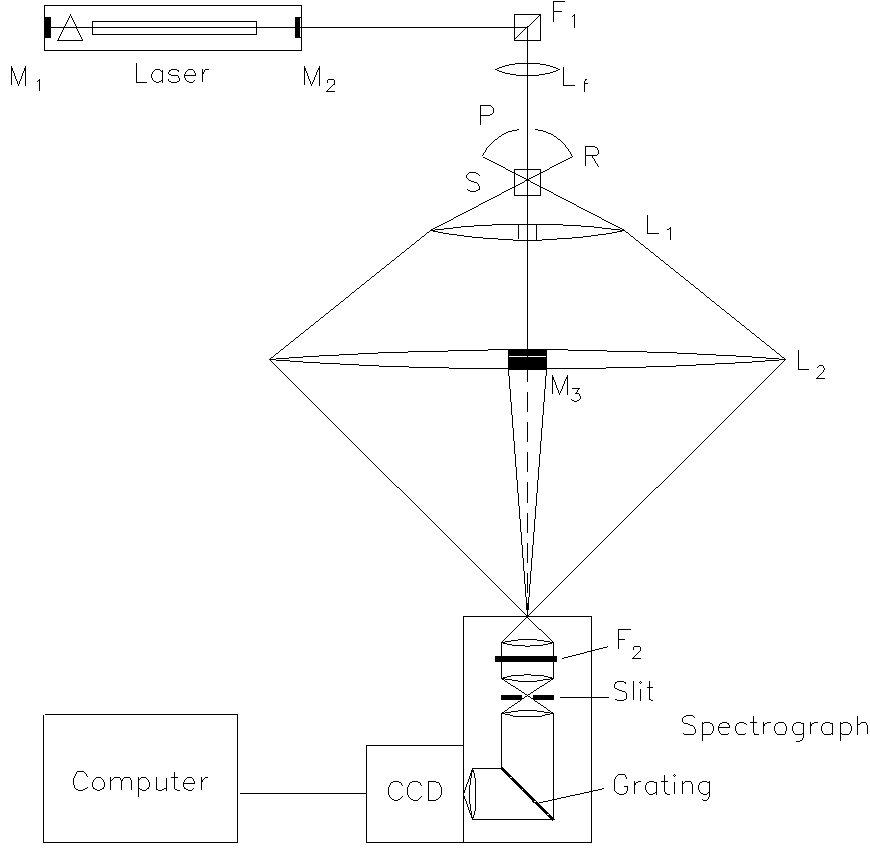
Figure 1. The experimental set-up
The excitation unit consists of an argon ion laser (Coherent, model Innova 90), a band pass filter (F1; Kaiser optics, model HLBF-488-1.0), a focusing lens (Lf), a pinhole (P), and the feedback mirror (M3). The laser is set to a wavelength of 488 nm at a power of 3 W. After passing the sample cell (S), the laser beam is coupled back into the laser by the feedback mirror (radius 60 cm, reflectivity 99.9%). This enhances the laser power to a maximum value of up to 8 W inside the sample. To achieve this increase in power, the feedback mirror must be adjusted in such a way that its radius of curvature fits the radius of the phase front of the laser beam. Compared with a simple intracavity setup, as it has been used mainly for the measurement of gaseous samples [7], the coupled resonator has the advantage that the alignment of its different optical components can be more easily optimized. Furthermore, even if problems arise, e.g., by the formations of thermal lenses inside the sample, Raman spectra can still be recorded at the lower output power of the resonator formed by the mirrors M1 and M2.†
† Editor’s Note: The technique sounds more difficult than it is, so follow Hans’ advice and try it. We used it at Southampton as early as 1970 to record the rotational Raman of Cl2 see Spectrochim. Acta. 28A 1949 (1991). One day I must write a piece for you on rotational Raman. Trouble is it is not vibrational spectroscopy!!
The bandpass filter F1, which consists of a holographic diffraction grating between two quartz prisms, reduces the spontaneous emission lines from the argon discharge. This task is achieved by effectively directing the exciting laser beam at an angle of 90 degrees. The spectral lines, that contaminate the monochromatic laser radiation are angularly dispersed and further rejected by a pinhole (P). The hole (diameter 3 mm) is drilled into a reflector of stainless steel ®. The reflector R is also part of the light collecting unit. The lens Lf produces a sharp focus of the laser beam with a diameter of about 10 µm in the sample.
The sample cell S has an optical path length of 1 cm and a width of 4 mm. Filling the cell with 1 ml of liquid leads to a column height of about 2.5 cm. This avoids an unnecessarily large cutoff of the scattered light, since the laser beam is focussed inside the cell at a height of 1 cm.
With regard to HPLC; our sample cell can be replaced by a flow cell. An HPLC column (LichroCART 125-4 with Lichrospher 100 RP 18 (Merck)) was used in combination with the appropriate precolumn – LichroCART 4-4. The selected injection volume was 100 µL for each measurement. The flow velocity of the mobile phase (water/acetonitrile 50:50) was set to the commonly selected value of 1 mL/min.
The light collection unit (magnification <10, depending on its alignment) consists of two aspheric multi-element lenses (“Fresnel lenses” from LOT-Oriel), made of acrylic plastic, and a spherical reflector (R). The first lens (L1, focal length 7.6 cm, diameter 15 cm) has a hole of 3 mm diameter in its centre to let the laser beam pass without focusing. The second lens (L2, focal length 45.7 cm, diameter 45 cm) holds the feedback mirror M3, which is fitted into a bore in its centre. This construction reduces the shadowing of the scattered light by the mirror and its holder to a minimum. Furthermore, that part of the light which is scattered in the reverse direction of the imaging lenses is collected by reflector R (radius of curvature 2.5 cm, diameter 4 cm). The reflector is made of highly polished stainless steel and contains a hole (P) for the laser passage.
The use of multi-element Fresnel lenses has several advantages. They are excellent for imaging a pointlike light source into a pointlike image, i.e. for illuminating the entrance slit of a spectrometer. They give better optical apertures (ratios of focal length to diameter), hence larger solid angles of scattered light can be collected and they are better corrected for spherical aberration than single element lenses. Their large diameter diminishes the amount of scattered light lost by the holes in their centres. Finally, since Fresnel lenses are lighter than conventional ones of comparable optical properties, their adjustable mountings easier to construct.
The light detection unit contains the spectrograph (Kaiser optics, model HoloSpec f/1.8i) and the CCD camera (Photometrics, model SDS 9000 with a head CH 270). This spectrograph has an excellent entrance optical aperture (f/1.8) that ensures that practically all the light passing the slit is imaged onto the focal plane. Inside the housing of the spectrograph, a holographic notch filter (SuperNotch Plus model HSPF-488AR-2.0) is inserted between the entrance plane of the spectrograph and the internal slit (SL, 100µm). This filter suppresses the Rayleigh scattering that would generate a large amount of stray light. The reduction of stray light enhances the signal to background ratio considerably.
The camera is cooled with liquid nitrogen (temperature adjusted to -90° C) and equipped with a 1024×256 pixel CCD chip (Metachrome™ II extended UV). Its sensitivity is high enough to detect signals of only a few photons. The spectral range of the setup encloses about 3000 cm-1 from the excitation line, and the achieved spectral resolution in the central region of the chip is about 10 cm-1. This corresponds to illuminating three pixels.
The scattering zone which contributes principally to the light that passes the entrance slit of the spectrograph is very small. It is concentrated in a cylinder of about 10µm diameter and a length of about 1 mm. Most of the scattered light produced from the volume of the diverging beam and outside this zone does not enter the spectrograph. However calculation of the total detected amount of light from the beam in the sample as a function of this cylinder length suggests that the cylinder contributes about two thirds of the available signal. Hence, the main part of the scattered light originates from a sample volume of less than 1 nL.
HPLC measurements start by recording signals from the UV/VIS detector. Once an interesting peak is visible in the chromatogram, the eluate is purged into the Raman sample cell. Otherwise the eluate is directed through a bypass. HPLC peaks typically have a full width of about one minute (with a FWHM of about 20 to 30 s), and provides the required amount of about 1 mL of liquid to fill the sample cell at typical flow rates. In the case of more than one interesting peak in the HPLC spectrum, the samples are collected and passed into the sample cell successively.
The sensitivity of the measurements of the CCD is limited by the amount of background signal. This depends on the number of pixel columns in the selected pixel area and on the total number of incoming photons in the same area. To obtain best results, the spectral range must be set to a region which is characteristic of the examined molecules and not congested by the Raman signals of the solvent. To avoid saturation effects on the CCD, the total measuring time (typically 150 s) is divided into a sequence of several single exposures (typically 10 to 50 s each).
Near the detection limit, the evaluation of the data consists of several steps. The measured spectrum is corrected for background signals by subtracting a properly scaled spectrum of the pure solvent. The remaining background may contain fluorescence signals, which have a much larger spectral bandwidth than the Raman ones, and may show broad artificial structures that arise from imperfect correction. Both contributions to the background signal are removed to obtain a flat baseline. This is carried out by subtracting a carefully smoothed graph of the background. Finally, there are remaining periodic structures in the spectra caused by the CCD camera. These structures contribute a relatively large standard deviation to the Raman signal and the background and in this way limit the sensitivity of the measurements. The resulting spectrum is Fourier filtered.
We define the detection limit by the ratio of the height of Raman signals to the height of signals originated from adjacent residual structures. The detection limit is reached, when the ratio is unity.
Results
The ability of the setup to detect small sample concentrations was first tested by measuring dilute solutions of benzene. Acetonitrile was chosen as the solvent since it provides suitable spectral ranges in which vibrations of the solute are missing. Figure. 2, Figure 3 and Figure 4 present the most prominent Raman signal of benzene at 992 cm-1 in low-concentrated solutions. The peaks appearing between 1025 and 1250 cm-1 at lower concentrations are artificial and their behaviour is similar to noise. Restricting the measurement time for one spectrum to a value of about 150 s, the detection limit is around 3 10-6 mol/L. As can be seen from the number of counts in the spectra, the relation between the signal height and the selected concentrations is not linear. The non-linearity probably originated from both saturation effects of the CCD-chip and the effect of the mathematical procedure, we use to extract the spectropic data from the recorded spectra.
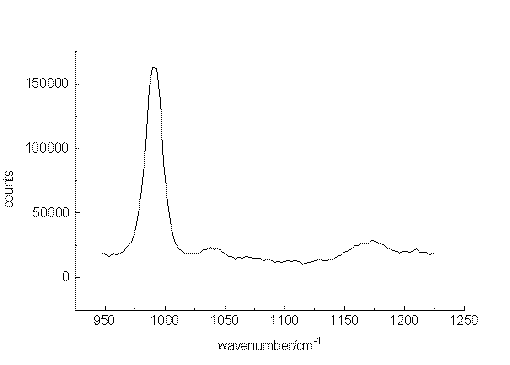
Figure 2. The Raman transition at 992cm-1 of benzene, solvent:acetonitrile, 10-2mol/L
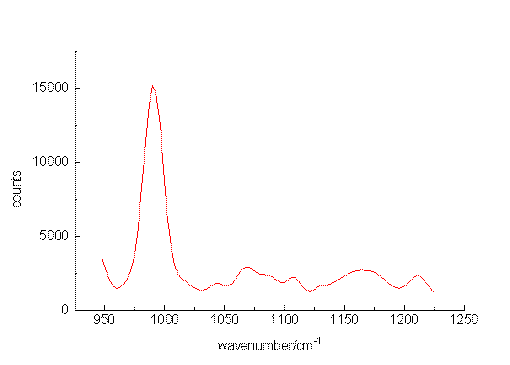
Figure 3. The Raman transition at 992 cm-1 of benzene, solvent: acetonitrile, 10-4mol/L
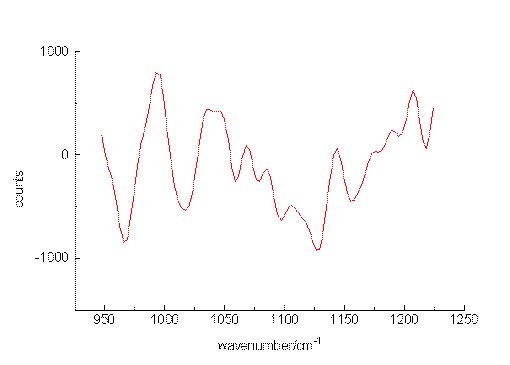
Figure 4. The Raman transition at 992 cm-1 of benzene, solvent: acetonitrile, 3 10-6 mol/L
As a second test, spectra were recorded from a mixture of m-xylene and p-xylene also diluted with acetonitrile. Figure. 5, 6 and 7 present Raman band transitions of p-/m-xylene mixtures at 10-2, 10-3 and at 10-5 mol/L within the range AV 950 and 1275 cm-1. At 10-2 mol/L, all Raman signals of the xylene isomers within the selected spectral range can be detected (Table 1). The small signals at about 1070, 1120 and 1130 cm-1 are artificial.
The xylenes could still be identified through their main bands at concentrations down to 10-5 mol/L (1µg/mL or 430 ppb) for each isomer. Here, the ratio of the signal heights to those of the noise is slightly larger than unity. Since the spectral position of the xylene signals remain constant, while the noise varies, an identification is possible at this very low concentration.
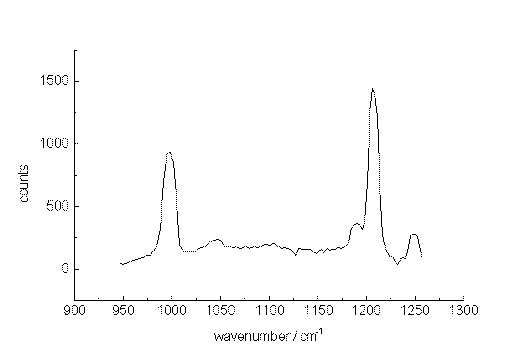
Figure 5. The Raman spectrum of p-xylene/m-xylene in acetonitrile at 10-2 mol/L between 950 and 1250cm-1
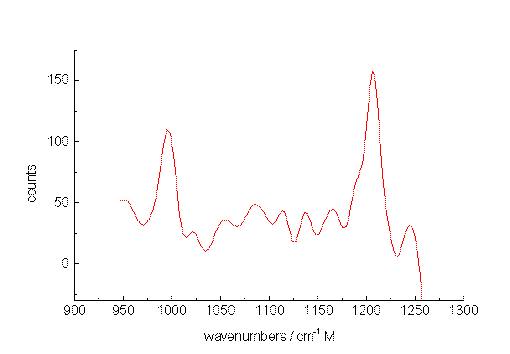
Figure 6. The Raman spectrum of p-xylene/m-xylene in acetonitrile at 10-3 mol/L between 950 and 1250cm-1
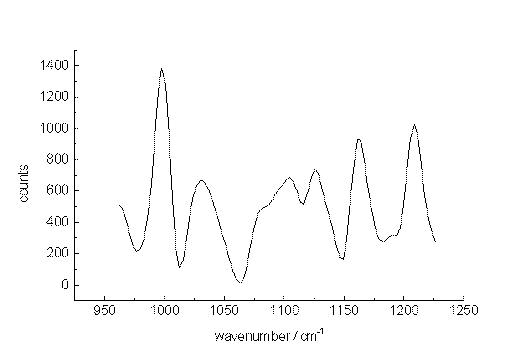
Figure 7. The Raman spectrum of p-xylene/m-xylene in acetonitrile at 10-5 mol/L between 950 and 1250cm-1
The final example demonstrates the efficiency of Raman spectroscopy in analysing multi-component systems. Figure. 8 shows Raman spectra of the dichlorophenols isomers each at 10-4 mol/L dissolved in acetonitrile as well as the spectrum of the mixture including all dichlorophenols. The dichlorophenols differ only in the substitution pattern of the two chloroatoms. Since their chromophores are almost identical, the electronic spectra (both absorption and fluorescence spectra) of the isomers are nearly indistinguishable. On the other hand, their Raman spectra are very different. Without going into details it is quite clear from the bottom spectra of P.8 that all the isomers can easily be identified.
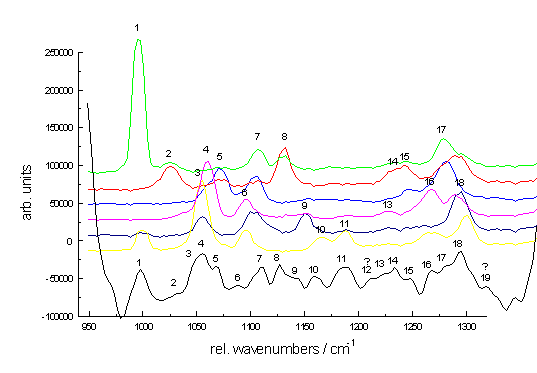
Figure 8. Raman spectra of the dichlorophenols between 950 and 1350 cm-1; the Raman peaks of all spectra are numbered consecutively; the graph at the bottom presents the collective spectrum;
spectra of single compounds: green: 3,5-dichloro, red: 3,4-dichloro, blue: 2,6-dichloro, magenta: 2,5-dichloro, dark blue: 2,4-dichloro, yellow: 2,3-dichloro
| isomer | wavenumber/cm-1 | Wilson’s number | assignment |
| m-xylene | 9981039
1095 1171 (sh) |
12-
18a 9b |
C-Cring stretch, C-Cring in-plane bendingC-Hmethyl bending
C-Cring stretch, C-C-H in-plane bending C-C-H in-plane bending |
| p-xylene | 1183 (sh)1206 | 9a7a | C-C-H in-plane bendingC-Cring stretch |
TABLE 1. Raman transitions of m-xylene and p-xylene partially visible in Figures. 5 and 6. The assignments are made due to [8]
Now we the authors are spectroscopists! The examples we present only show the possibilities of solving problems in trace analysis by Raman spectroscopy and of combining HPLC with Raman spectroscopy. We would be grateful to receive suggestions concerning real analytical chemical problems from readers of this article.
Considerably lower concentrations leading to the analysis of real traces could be attempted if we use resonance effects. Our experimental setup needs little alteration in Raman scattering to work in the ultraviolet range, where most molecules show resonance. The only disadvantage arises from the fact that large multi-element lenses made of quartz are not available. As a result, standard lenses must be used. This will cause some loss in sensitivity but the enhancement due to resonance will improve the overall sensitivity of the Raman spectrometer by a factor estimated to be a hundred or more.
References
- R. Steinert, H. Bettermann and K. Kleinermanns, Appl. Spectrosc. 51, 1644 (1997)
- K.Iriyama, Y.Ozaki, K.Hibi, and T. Ikeda, J. Chromatgr. 254, 285 (1983)
- M. D´Orazio and R. Hirschberger, Opt. Eng. 22, 308 (1983)
- H. Todori, and Y.A. Hirakawa, Chem. Pharm. Bull. 32, 193 (1984)
- A.P. Gamot, G. Vergoten, M. Saudemon, G. Fleury, and J. Barbillat, Talanta 33, 295 (1986)
- H. Koizumi and Y. Suzuki, J. High Resolut. Chromatogr.Chromatogr. Commun. 10, 173 (1987)
- A. Wehr, “High Resolution Rotational Raman Spectra of Gases”, in Raman Spectroscopy of Gases and Liquids, A. WeberEd.,Topics Curr. Phys., Vol 11 (Springer, Berlin, Heidelberg 1979), Chap.3
- L.M. Sverdlov, M.A. Kovner, and E.P. Krainov, Vibrational Spectra of Polyatomic Molecules, (John Wiley and Sons, New York, Toronto, 1974)