Mapping the crystalline morphology of isotactic polypropylene by infrared microscopy
G. Ellis*, M. A. Gómez and C. Marco
Departamento de Física e Ingeniería,
Instituto de Ciencia y Tecnología de Polímeros,
CSIC, c/ Juan de la Cierva 3,
28006 Madrid,
Spain.
*Email: gary [at] ictp.csic.es
Abstract
Infrared microscopy has been used to study the crystalline morphology of two polymorphs of isotactic polypropylene. Some of the minor differences observed between the mid-infrared spectra recorded from a– and b-iPP have been used to generate a structural image of a sample containing both a– and b-spherulitic morphology.
Introduction
One of the most important aspects of semicrystalline polymers is the crystalline morphology, since the ultimate properties of the material depend fundamentally on microstructural factors, such as the degree of crystallinity, the size and type of spherulites formed, the lamellar thickness and the crystalline orientation. Isotactic polypropylene, iPP has a particularly complicated crystalline microstructure, which depends, amongst other factors, on the mechanism and rate of crystallisation. Four different crystalline structures have been described [1], corresponding to monoclinic (a), hexagonal (b), triclinic (g), and smectic or quenched polymorphs, the monoclinic structure being that principally obtained under typical industrial and laboratory processing conditions. The b-form occurs more rarely than the a-form since it is thermodynamically less stable, although under given thermal conditions it can occur simultaneously [2], and its appearance can be favoured under conditions of shear stress [3]. Techniques available for the preparation of samples with controlled levels of the b-modification are principally the temperature gradient method [2,4], and the use of b-nucleating agents [2], the latter being of particular interest in the polymer industry.
Although there are numerous vibrational spectroscopic studies of polypropylene in the literature, only a handful of studies have compared the different crystalline morphologies in iPP. Both far-infrared [5,6] and Raman [7,8] studies have suggested that the small variations observed in the spectra of various polymorphs can be associated with the different environments in which the polymer chains are found [9,10], and must be associated with the packing geometries and unit cell parameters. The most relevant variations appear to be associated with the pendant methyl group, which is clearly the most susceptible to intermolecular interactions.
In the present work, the mid IR spectra of a-iPP and b-iPP are presented with the aim of spectroscopically differentiating between the polymorphs.
Experimental
A commercial grade of isotactic polypropylene with an isotacticity index of 95% was kindly supplied by REPSOL-YPF, and its physical characteristics are described elsewhere [11]. Two nucleating agents at a concentration of 0,05% by weight, 1,2,3,4-bis-(3,4-dimethylbenzylidene sorbitol), MILLAD 3988 from Milliken, and N,N’-dicyclohexyl-2,6-naphthalate dicarboxamide, NJSTAR NU100 from NJC Rika, were employed in order to generate predominantly a-iPP and b-iPP respectively.
Film samples for IR microscopy were prepared in two steps. Firstly, films of varying thickness between 30 – 150 mm were prepared in a hot-press by melting at 210ºC for 5 minutes at a pressure of 100 Bar, and rapidly cooling between metal plates at 15ºC. Secondly, sections of the resulting films were carefully selected and mounted on a microscope slide beneath a cover slip, and introduced into a Mettler FP80-HT microscope hot stage. After melting the films at 210ºC for 10 minutes in order to remove any previous thermal history, the iPP samples were crystallised either (A) dynamically to room temperature at a cooling rate of 10ºC/min, or (B) isothermally by cooling at 20ºC/min to a temperature of 130ºC where the samples were held for 60 minutes, before cooling rapidly to room temperature. The development of the crystalline morphology in the latter case was monitored through crossed Polaroids in a Reichert Zetopan-Pol polarising optical microscope. The samples, summarised in Table 1, were then carefully separated from the microscope slides and adequately mounted for transmission infrared microscopy.
|
Sample |
Type of nucleating agent |
Thermal history |
Dominant crystalline form |
|
1 |
none |
A |
a |
|
2 |
a |
A |
a |
|
3 |
b |
A |
b |
|
4 |
none |
B |
a |
Table 1. Description of samples
Infrared spectra were recorded at a resolution of 4 cm-1 using a Perkin Elmer i-Series IMAGE microscope fitted with Polaroid filters, coupled to a System 2000 FTIR spectrometer, using the aperture sizes specified in the figures. Perkin Elmer IMAGE and Spectrum software was used to analyse the data.
Wide angle X-ray diffractograms, WAXS, of samples 1-3 were obtained at room temperature using a Phillips Geiger counter X-ray diffractometer, at 1º/min over a 2qrange between 5 – 35º using Ni-filtered CuKa radiation.
Results and Discussion
Figure 1 shows the WAXS diffractograms obtained from pure iPP and a– and b– nucleated iPP crystallised dynamically at 10ºC/min, where it can be clearly observed that both samples 1 and 2 show the characteristic crystalline reflections of iPP in its a-modification, at 14.2º, 17.0º, 18.8º, 21.2º and 22.0º, whereas sample 3 shows two sharp reflections at 16.2º and 21.2º along with other weaker reflections, characteristic of the b-modification [2]. The very high concentration of b-iPP, around 90%, generated by this nucleating agent has been previously described [12].
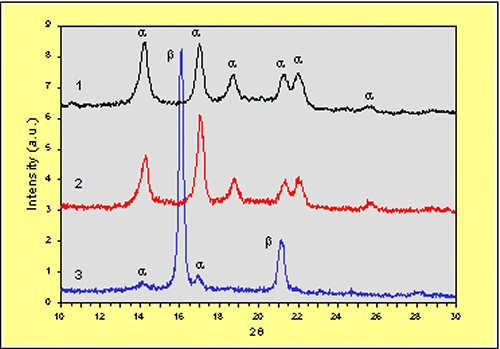
Figure 1. X-ray diffractograms of samples 1-3.
The infrared spectra are presented in Figure 2. Apart from two small bands which are detected in samples 2 and 3 at around 875 and 1630 cm-1, and correspond to the additives MILLAD 3988 and NJSTAR NU100, respectively, the differences between the spectra of iPP are much more subtle, observing only very small wavenumber shifts and intensity changes in a number of bands, illustrated in the inset for the region between 1340 – 1200 cm-1. This is a reasonable observation, since the conformation of the chains in each modification is identical, corresponding to a 31helix with identical intramolecular interaction energies [1]. However, analysis of these minor spectral variations, which are described in more detail elsewhere [13], have allowed us to systematically differentiate between a– and b-iPP.
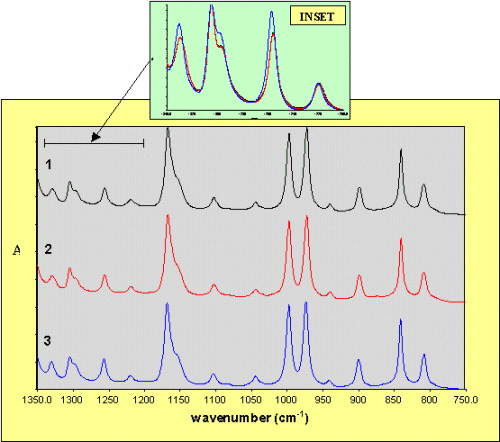
Figure 2. Infrared spectra of samples 1-3,
recorded with an aperture of 100mm.
For sample 4, isothermal crystallisation at 130ºC results in a much lower crystallisation rate, and consequently larger and more ordered spherulites can be formed. Figure 3 shows a visible image survey of a region of this sample observed in the infrared microscope using polarised visible light. It can be seen that both polymorphs have crystallised simultaneously, and although the sample contains mainly a-iPP, a small amount of b has been formed. In the area studied a single b-spherulite is located amongst a well-developed a-spherulitic matrix.
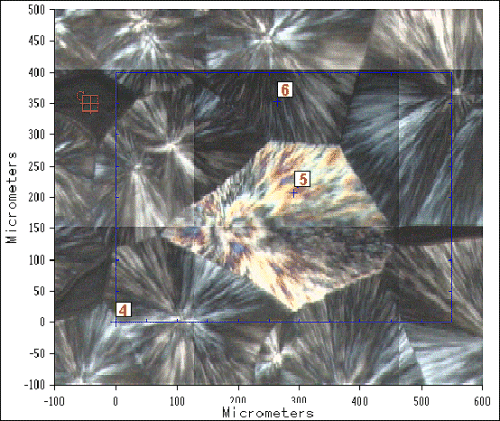
Figure 3. Polarised visible image survey or part of sample 4,
recorded in the infrared microscope.
Spectra recorded at the positions marked on the figure were characteristic of the a– and b-modifications, showing similar variations as those described for the nucleated systems some of which are illustrated in Figure 4.
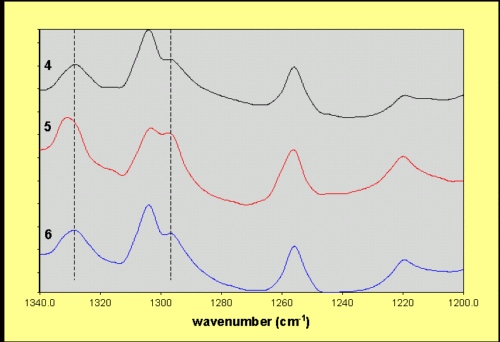
Figure 4. Spectra recorded from sample 4
in the positions marked 4, 5 and 6 on Figure 3.
An area of 400 x 550 mm described by the blue box in Figure 3 was studied by point-by-point mapping using a square aperture of 25 mm (shown), recording spectra at 25 mm steps. By considering the relative absorbance of a number of bands, it has been possible to generate maps of the sample that are characteristic of the crystalline morphology. As an example, Figure 5 shows the 2D band ratio contour map and the corresponding 3D axonometric projection, for the relationship between the integral areas of the band at around 1330 cm-1 corresponding to CH2 bending and twisting modes [14-16], and those at 1168 and 1152 cm-1, corresponding to combination bands which contain principally nCC chain stretching modes along with significant contributions from nC-CH3 stretching and gCH3 rocking modes [14,15].
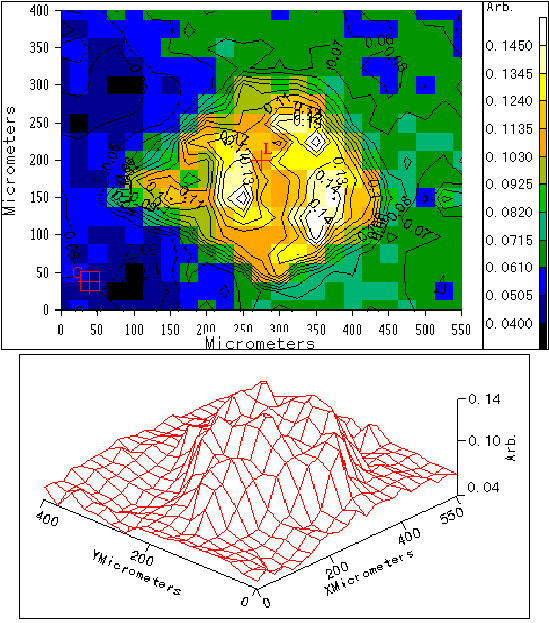
Figure 5. Band ratio contour map and
axonometric projection of sample 4.
It should be noted that the correlation between the polarised optical micrograph and the IR structural maps is very good, and from the significant differences in the relative intensities of the IR bands we are able to clearly distinguish between the two crystalline morphologies.
This type of observation has been previously reported for poly(vinylidene fluoride), where a specific band which only appears in the g-modification was used to map a small g-spherulite embedded in a larger a-spherulite [17]. Although the case for polypropylene is more complex, correlations similar to those used to generate Figure 5 can be made with other bands, and are not exclusively associated with the methyl group vibrations [13].
Finally, it should be pointed out that careful control of the samples is essential, since many bands in iPP are affected by the thermal and mechanical history. Moreover, variations in the crystallinity of the samples can also affect the data, and although this is not the most important effect in the present case [13], crystallinity differences between a– and b-iPP crystallised under the same conditions are commonly observed.
Conclusion
The subtle variations that occur between the IR spectra of the a– and b-crystalline modifications of iPP can be exploited to distinguish between the two polymorphs, and by applying these spectral differences using IR microscopy it is possible to generate structural maps of the crystalline morphology over defined sample areas. This type of study may be important in the development of heterogeneous iPP systems, since the occurrence, nature and distribution of the different polymorphic structures conditions important final properties of the materials such as transparency and modulus.
Acknowledgements
The authors acknowledge financial support from the following research projects: CICYT Mat-98-0914 and CAM-07N/0032/1999.
References
- S.Z.D. Cheng, J.J. Janimak, J. Rodríguez, in “Polypropylene. Structure, blends and composites. I. Structure and morphology”, J. Karger-Kocsis (ed.), Chap.2, p.31, Chapman & Hall, London (1995)
- J. Varga, in “Polypropylene. Structure, blends and composites. I. Structure and morphology”, J. Karger-Kocsis (ed.), Chap.3, p.56, Chapman & Hall, London (1995)
- J. Varga, J. Karger-Kocsis, Polym. Bull., 30, 105 (1993)
- J.M. Crissman, J. Polym. Sci., A2, 389 (1969)
- M. Goldstein, M.E. Seeley, H.A. Willis, V.J.I. Zichy, Polymer, 14, 530 (1973)
- D.R. Beckett, J.M. Chalmers, M.W. Mackenzie, H.A. Willis, H.G.M. Edwards, J.S. Lees, D.A. Long, Eur. Polym.J., 21, 849 (1985)
- S.D. Merajver, S.L. Wunder, W. Wallace, J. Polym. Sci. Polym. Phys. Ed., 23, 2043 (1985)
- J.M. Chalmers, H.G.M. Edwards, J.S. Lees, D.A. Long, M.W. Mackenzie, H.A. Willis, J. Raman Spectrosc., 22, 613 (1991)
- A. Bunn, M.E.A. Cudby, R.K. Harris, K.J. Packer, B.J. Say, Polymer, 23, 694 (1982)
- M.A. Gómez, H. Tanaka, A.E. Tonelli, Polymer, 28, 2227 (1987)
- C. Marco, M.A. Gómez, G. Ellis, M.J. Arribas, J. Appl. Polym. Sci. (2001) in press
- C. Marco, M.A. Gómez, G. Ellis, M.J. Arribas, J. Appl. Polym. Sci. (2001) in press
- G. Ellis, M.A. Gómez, C. Marco, Polymer (2001) in press
- T. Miyazawa, Y. Ideguchi, K.Fukushima, J. Chem. Phys., 38, 2709 (1963)
- R.G. Snyder, J.H. Schachtschneider, Spectrochim. Acta, 20, 853 (1964)
- H. Tadokoro, M Kobayashi, M. Ukita, J. Yasufukum, S. Murashashi, J. Chem. Phys., 42, 1432 (1965)
- J. Kressler, R. Schäfer, R. Thoman, Appl. Spectrosc., 52, 1269 (1998)
Received 16th July 2001, received in revised format 10th September 2001,
accepted 10th September 2001.
REF: G. Ellis, M. A. Gómez and C. Marco Int. J. Vib. Spect., [www.irdg.org/ijvs] 5, 4, 7 (2001)