Estimation of the P-I bond length in PI4+AsF6–by Raman and 31P MAS NMR Spectroscopy
Christoph Aubauer and Thomas M. Klapötke*
Department of Chemistry,
Ludwig-Maximilians-University,
Butenandtstr. 5-13 (D),
D-81377 Munich, Germany,
*To whom correspondence should be addressed
Correspondence e-mail: thomas.m.klapoetke [at] cup.uni-muenchen.de
Keywords
tetrahalophosphonium cations,
31P MAS NMR spectroscopy, Raman spectroscopy
Abstract
The P-I bond length in almost isolated PI4+ salt PI4+AsF6– is estimated, based on the results of vibrational and solid-state 31P MAS NMR spectroscopic studies of several tetraiodophosphonium salts containing AlI4–, GaI4–, AlBr4–, AlCl4– and AsF6– and the X-ray crystal structure analyses of the tetraiodophosphonium salts PI4+GaI4–, PI4+AlI4–, PI4+AlBr4– and PI4+AlCl4–. Influences of the counter-ion on the 31P chemical shift and the vibrational frequencies are discussed.
Introduction
Recently [1], we showed in a combined theoretical and experimental study, that the PI4+ cation has an extremely large negative 31P chemical shift in the compounds PI4+AsF6– (d = – 519 ppm) and PI4+SbF6–(d = – 517 ppm), which is due to spin-orbit (SO) contributions from the four heavy iodine substituents, transmitted to the phosphorus nucleus by a very effective Fermi-contact mechanism. The less negative solid-state 31P NMR chemical shifts found in the PI4+AlI4– with strong [PI4]+…[AlI4]– interactions (d=– 305 ppm) and PI4+GaI4– (d = – 295 ppm), suggest that the P-I bond orders are reduced due to intermolecular I × × × I interaction between PI4+ cations and EI4– (E = Al, Ga) anions [1].
Moreover, the solid-state 31P MAS NMR studies for PI4+ species showed that the 31P isotropic shifts ranging from d = – 295 to – 519 ppm depend considerably on the nature of the counter-ion (Figure 1) [1, 2]. The more isolated the character of the PI4+ cation (shorter P-I bond lengths, weaker cation × × ×anion interactions), the larger becomes the P-I bond order. Consequently, the Fermi-contact mechanism that transfers the spin-orbit induced spin density to the phosphorus nucleus is more efficient [3], and the 31P resonance is shifted to higher field. Significant donor-acceptor contacts between cation and anion in the solid-state lead to less pronounced low frequency isotropic 31P shifts [1, 2].
Additionally, a trend towards higher wave numbers for the fundamental frequencies of PI4+ could be observed from the polymeric [PI4+EI4– (E = Al, Ga)] to the “ideal” ionic cases [PI4+MF6– (M = As, Sb)] [1, 2, 4].
Based on the results of the vibrational and 31P MAS NMR results and the X-ray crystal structure analyses of the tetraiodophosphonium salts PI4+GaI4–[2], PI4+AlI4– [5], PI4+AlBr4– [2] and PI4+AlCl4– [2] we report in this paper the estimation of the P-I bond length in the not yet structurally characterised salt PI4+AsF6–, which contains the almost isolated PI4+ cation and was first reported in 1990 [6].
Results and Discussion
Table 1 shows a significantly decrease of the average P-I bond length in the PI4+unit going from PI4+GaI4– (2.408(4) Å) to PI4+AlCl4– (2.368(4) Å) with an opposite trend for the interatomic I × × × X distances (X = I, Br, Cl; PI4+GaI4– : 3.357(2) – 3.430(2) Å; PI4+AlCl4–: 3.315(8) – 3.511(3) Å; Table 1) [2]. The molecular structures of PI4+AlI4– and PI4+GaI4– show rather short interatomic I × × × I between the PI4+ and the EI4– units (E = Al, Ga), which are significantly shorter than the sum of van der Waals radii (ca. 4.30 Å [7]), indicating strong cation × × × anion interactions, while the crystal structures of PI4+AlBr4– (3.380(2) – 3.449(2) Å, sum of van der Waals radii: ca. 4.10 Å [7]) and PI4+AlCl4–(3.315(8) – 3.511(3) Å, sum of van der Waals radii: ca. 3.95 Å [7]) show considerably weaker interatomic I × × × X contacts (X = Br, Cl), displaying a more isolated character for PI4+ in this species.
|
Table 1. 31P chemical shifts, n 1 (A1, PI4+) stretching vibrations, average P-I bond lengths and interatomic cation × × × anion distances for some PI4+ species |
|||||
| PI4+GaI4–a,b,c | PI4+AlI4–a,c,d |
PI4+AlBr4–b | PI4+AlCl4–b | PI4+AsF6–a,c | |
| d 31P NMR [ppm] |
– 295 |
– 305 |
– 416 |
– 456 |
– 519 |
| n 1 (A1, PI4+) [cm-1] |
151 |
152 |
165 |
169 |
178 |
| Æ d (P-I) [Å] |
2.408(4) |
2.396(9) |
2.381(4) |
2.368(4) |
|
|
d (I × × × X) [Å] X = Cl, Br, I |
3.357(2) – 3.430(2) |
3.386(4) – 3.451(3) |
3.380(2) – 3.449(2) |
3.315(8) – 3.511(3) |
|
|
a see ref. [1]; b see ref. [2]; c see ref. [4]; d see ref. [5] |
|||||
|
P-I bond strength ←—————————————————- cation × × × anion-interactions |
|||||
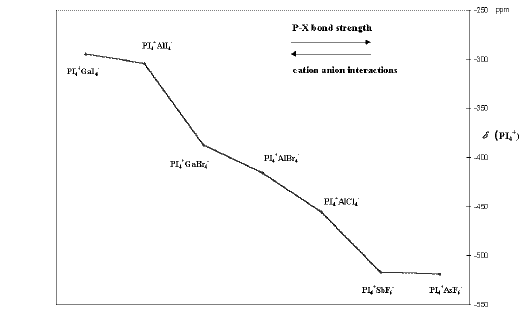
Figure 1. 31P NMR chemical shifts for some PI4+ species.
Figure 1 summarises the 31P NMR chemical shifts of some PI4+ species in the presence of different counter-anions. The isotropic chemical shift of PI4+ is dependent on the nature of the counter-anion and varies in a considerably large range, between d = – 295 ppm (PI4+GaI4– , Table 1) and d = – 519 ppm (PI4+AsF6– , Table 1) [1, 2]. Significant bridging cation × × × anion interactions in the lattice result in a weakening of the P-I bonds in PI4+. The more isolated the cation, the more efficient is the Fermi-contact mechanism that transfers the SO-induced spin density to the 31P nucleus, 31P resonance is shifted to low frequency. Thus, it is possible to take the low frequency 31P chemical shift as a measure of the intermolecular interactions.
The Raman spectra of PI4+GaI4– , PI4+AlBr4– and PI4+AlCl4– are shown in Figure 2. The vibrational normal modes of the PI4+ cation in PI4+GaI4– , PI4+AlI4– , PI4+AlBr4– , PI4+AlCl4– and PI4+AsF4– are summarised in Table 2 [1, 2, 4]. The Raman-active symmetric n 1 (A1) stretching mode of PI4+ species can be observed as the most intense peak in the Raman spectra. Analogous to the isotropic 31P chemical shift of the PI4+ cation, it is a characteristic frequency to indicate the extent of intermolecular cation × × × anion interactions in these species.
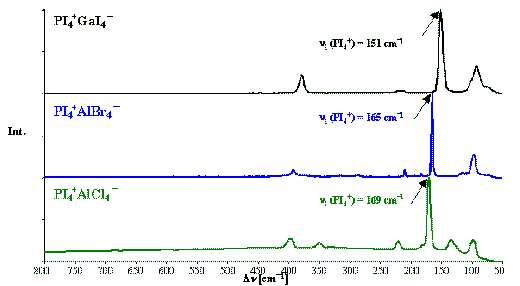
Figure 2. Raman spectra of PI4+GaI4– , PI4+AlBr4– and PI4+AlCl4–
|
Table 2. Experimentally observed fundamental Raman frequencies for some PI4+ species |
|||||
|
assignment |
PI4+GaI4–a,c |
PI4+AlI4– |
PI4+AlBr4–b |
PI4+AlCl4–b |
PI4+AsF6–a,c |
|
n 3 (T2, PI4+) |
378 |
380 |
392 |
400 / 394 |
|
|
n 1 (A1, PI4+) |
151 |
152 |
165 |
169 |
178 |
|
n 4 (T2, PI4+) |
94 |
95 |
98 |
98 |
82 |
|
n 2 (E, PI4+) |
72 |
77 |
71 |
71 |
|
|
a see ref. [1]; b see ref. [2]; c see ref. [4]. |
|||||
Due to decreasing average P-I bond lengths (increasing P-I bond order, diminishing cation × × × anion interactions), the symmetric n 1 (A1) stretching vibration of the PI4+ salts move increasingly to higher wave numbers along the series PI4+GaI4– (151 cm–1) < PI4+AlI4– (152 cm–1) < PI4+AlBr4– (165 cm–1) < PI4+AlCl4– (169 cm–1) < PI4+AsF6– (178 cm–1, Table 1 and 2).
In the case of PI4+AsF6– the shift to higher wave numbers (n 1 (A1) = 178 cm–1) is largest. This is consistent with the well-known non co-ordinating character of AsF6– anions, and with the expectation that no significant interactions between cations and anions occur for this system.
To summarise, the increasing P-I bond order (shorter P-I bond lengths, stronger P-I force constants) in the PI4+ cation causes a low frequency shift of the 31P chemical shift (d 31P (PI4+): – 295 to – 519 ppm) and a shift to higher wave numbers of the normal modes of PI4+ in the vibrational spectra (e.g. n 1 (PI4+): 151 to 178 cm–1) from the PI4+ salts PI4+EI4– (E = Al, Ga) with strong [PI4]+…[EI4]– interactions to the almost isolated PI4+ salt PI4+AsF6– (Table 1).
In Figure 3 the average P-I bond distance dependent on the 31P chemical shift and the symmetric n 1 (A1) stretching mode of the PI4+ cation is represented. The linear regression of both sets of experimental values [n 1 (PI4+) and d(PI4+)] indicate that the average P-I distance in PI4+AsF6– can be estimated to lie in the range between 2.350 and 2.354 Å.
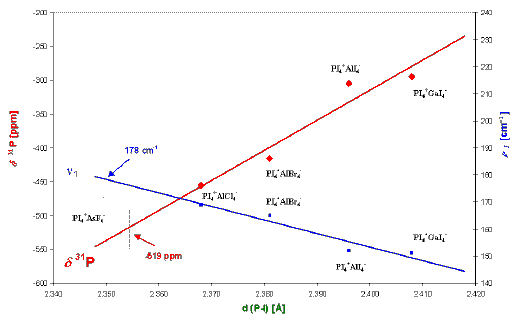
Linear regression: u for d 31P (PI4+): d (P-I) = 1/10991 [d 31P (PI4+)] + 2.47
n for n 1 (PI4+): d (P-I) = – 1/502 [n 1 (PI4+)] – 2.7
® dcalc. (P-I) » 2.350 – 2.354 Å
Figure 3. Estimation of the average P-I bond length
in PI4+AsF6 by linear regression.
In an earlier study the PI4+AsF6– Raman frequencies for the PI4+ cation were found at 193, 89 and 71 cm–1 [6] (Table 3). These values agree moderately with the present findings (Table 2 and 3). The quantum chemically attempted values have also been included into Table 3.
|
Table 3. Experimentally observed Raman frequencies of the PI4+ cation (AsF6–salt) and computed values. |
|||
|
assignment |
ref. [6] |
ref. [4] |
R MPW1PW91 a) |
|
n 1 (A1, PI4+) |
193 |
178 |
180 |
|
n 2 (E, PI4+) |
71 |
71 |
64 |
|
n 3 (T2, PI4+) |
– |
– |
419 |
|
n 4 (T2, PI4+) |
89 |
82 |
99 |
|
d / Å |
2.378 |
||
|
zpe / kcal mol– 1 |
2.67 |
||
| a cc-pVQZ basis set for P and ECP-MDF-46 for I with a 7s7p2d1f/3s3p2d1f valence basis set; – E = 386.949738 a.u.. | |||
Conclusion
In conclusion, the experimental results [1, 2, 4] (X-ray crystal structure analyses, vibrational spectroscopy, solid-state 31P NMR MAS spectroscopy) show a significant influence of the counter-ion on the 31P chemical shift and the vibrational frequencies with a linear relation between the P-I bond distance and the symmetric n 1 (A1) stretching mode and the isotropic 31P chemical shift, respectively, in these PI4+ species. Moreover, the average P-I distance in the almost “ideal” ionic PI4+ salt PI4+AsF6– can be estimated by linear regression to lie in the range between 2.350 and 2.354 Å.
Acknowledgement
We gratefully acknowledge the support of the Fonds der Chemischen Industrie and the University of Munich.
References
- M. Kaupp, Ch. Aubauer, G. Engelhardt, T. M. Klapötke, O. L. Malkina, J. Chem. Phys., 110, 3897 (1999).
- Ch. Aubauer, M. Kaupp, T. M. Klapötke, H. Nöth, H. Piotrowski, W. Schnick, J. Senker, M. Suter, J. Chem. Soc., Dalton Trans., 1880 (2000).
- M. Kaupp, O. L. Malkina, V. G. Malkin and P. Pyykkö, Chem. Eur. J., 4, 118 (1998).
- Ch. Aubauer, T. M. Klapötke, A. Schulz, Internet J. Vibr. Spec.[www.irdg.org/ijvs], 3, 2, 6 (1999).
- S. Pohl, Z. Anorg. Allg. Chem., 498, 15 (1983).
- I. Tornieporth-Oetting and T. M. Klapötke, J. Chem. Soc., Chem. Commun., 132 (1990).
- “Handbook of Chemistry and Physics“, 52. ed, ed. R. C. Weast, The Chemical Rubber Co., Cleveland, 1971-1972, p. D-146.
Received 26th June 2001, received in revised format 16th July 2001,
accepted 16th July 2001.
REF: C. Aubauer & T. M. Klapötke Int. J. Vib. Spect., [www.irdg.org/ijvs] 5, 4, 4 (2001)