Rapid Quantitative Analysis of Organophosphorus Pesticide Formulations by FT-Raman Spectroscopy
Stavroula G. Skoulika, Constantinos A. Georgiou* and Moschos G. Polissiou
Chemistry Laboratory,
Agricultural University of Athens,
75 Iera Odos, 11855 Athens,
Greece
*To whom correspondence should be addressed
Fax: +301-52 94 265,
Tel: +301-52 94 248
Email: cag [at] aua.gr
URL: www.aua.gr/georgiou
Abstract
The potential of Raman spectroscopy in the quantitative analysis of organophosporus pesticide formulations has been exploited using fenthion formulations as an example. The 2951, 1065, 661 and 604 cm-1 bands were used for calibration. Calibration curves based on the intensity and the area were linear (correlation coefficients: 0.991-0.998) in the concentration range of 0.4-4.36 M. Precision ranged from 0.4 to 6.8 % RSD (n=4) and detection limits were between 0.14 to 0.36 M. The 802 cm-1cyclohexane band was used for spectra normalization, resulting in improved long term stability of calibration curves. Results obtained during the analysis of a commercial formulation compare well with those obtained by the GC reference method. The proposed FT-Raman method is rapid, simple, safe, avoids sample pre-treatment and minimizes handling of toxic samples.
Keywords
Fenthion; FT-Raman; Determination; Pesticide formulations.
Introduction
The organophosporus insecticide fenthion (O,O-dimethyl-O-4-methylthio-m-tolyl phosphorothioate) has respiratory and contact action and is used for the control of bugs, pests, and insects [1]. For the quantitative analysis of fenthion in pesticide formulations several methods based on spectrophotometry after derivatization [2,3], gas chromatography [4] and infrared absorption [5] have been used. Spectrophotometric methods require extensive manual sample handling such as boiling and extraction. The gas chromatographic method is time consuming and the infrared absorption method requires sample dilution with carbon disulphide. As fenthion is a toxic cholinesterase inhibitor, alternative rapid analytical methods that overcome the aforementioned disadvantages are highly desirable.
Although Raman spectroscopy has found extensive use in structural and qualitative analysis, quantitative analysis has not kept pace with those applications. Thanks to multiplex measurements, FT-Raman spectroscopy achieves increased S/N ratio and high precision in frequency. The problems of fluorescence and thermal decomposition traditionally related to dispersive techniques are overcome with excitation at 1064 nm [6]. In comparison with IR methods, Raman analytical ones are less sensitive but are capable of measuring aqueous samples. Surface enhanced Raman spectroscopy (SERS) methods have been developed in order to increase sensitivity, but the achieved precision of 20-38 % RSD is poor [7,8].
In this work, the FT-Raman spectrum of fenthion is presented and a novel analytical method based on FT-Raman spectroscopy for the quantitative determination of fenthion in agrochemical organophosporus formulations is proposed.
Experimental
Sample preparation and chemicals
Bayer, Hellas kindly offered fenthion of 97% purity, xylene of technical grade, their fenthion formulation and two proprietary surfactants (MPS and DPS). The analytical fenthion standard of certified 99.1% purity was a kind offer from Bayer, Germany. All chemicals were used without further purification. While handling formulations and preparing standards, extreme care should be exercised to avoid fenthion contact with the eyes and skin. Fenthion is toxic, slightly soluble in water at 20o C (4.2 mg/l), with LD50 for mallard quail at 7.2 mg/kg [1]. The standard fenthion solutions were prepared by weighing the appropriate amount of fenthion and dissolving in xylene in the concentration range of 0.4-4.36 M.
Apparatus
FT-Raman spectra were recorded with a Nicolet 750 FT-Raman spectrometer equipped with a Nd:YAG laser source that emits at 1064 nm. An Indium-Galium Arsenide (InGaAs) detector, a CaF2 beamsplitter, and 180° back-scattering geometry were used. The laser beam was focused to the sample by a motorized positioner. For maximum optical efficiency, a manual side-to-side adjuster allows sample adjustment. To fine tune the spectrometer and maximize the detector signal, an optical bench alignment was performed before each batch of measurements. Wilmad WG-5M NMR tubes of 4.97 mm outer diameter and 0.38 mm wall thickness were used as sample cells. Spectra were accumulated from 100 scans collected during 3 min at a resolution of 4 cm-1. GC analysis was carried out using a Perkin-Elmer Sigma 3 equipped with a flame ionisation detector.
Results and discussion
Raman spectrum of fenthion
The Raman spectrum of fenthion is shown in Figure 1. Raman bands (Table I) are classified according to their % relative intensities to the 802 cm-1 cyclohexane band.
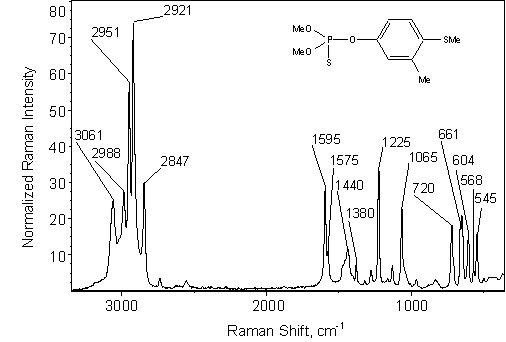
Figure 1. Fenthion Raman spectrum acquired at 1.01 w excitation intensity normalized to the 802 cm-1 cyclohexane band.
| Table I. Fenthion Raman bands (cm-1)a | |||||||
| Very strong | 2951 (58) | 2921 (74) | 1225 (34) | ||||
| Strong | 3061 (25) | 2988 (28) | 2847 (30) | 1595 (29) | 1065 (23) | ||
| Medium | 720 (18) | 661 (19) | 650 (21) | 604 (16) | 545 (16) | ||
| Weak | 1575 (11) | 1473 (7.2) | 1440 (12) | 1380 (9.6) | |||
| Very weak | 2737 (3.5) | 1278 (5.6) | 1162 (3.4) | 964 (3.3) | 831(3.3) | 568(6.2) | 499 (3.4) |
| a%Relative intensities to the 802 cm-1 cycloxehane band are shown in parentheses | |||||||
A clearly distinguishable Raman band between 600 and 700 cm-1 is characteristic for molecules possessing P=S bonds provided that they do not contain benzene rings [9]. For molecules that possess benzene rings, the P=S stretching is overshadowed by benzene ring vibrations [9, 10]. In fenthion spectrum two overlapping bands at 661 and 650 cm-1 appear in the aforementioned range. For the fenthion, additional strong bands at 1595 cm-1and a weak band at 568 cm-1 have been observed. The 831 cm-1 band could be assigned to mixed modes of predominantly P-(OR)2 stretch character [11]. The strong band at 1065 cm-1 could be attributed to the P-O-C bond stretch mode [10]. The intense bands appearing in the 2800-3100 cm-1 range are due to C-H stretching modes [9, 12-16].
Spectra normalization
Amongst other factors, Raman signals depend on excitation intensity, sample orientation and temperature variations [6]. As Raman spectroscopy is a single beam technique, these variations should be compensated during quantitative analysis. The use of an internal standard is the most obvious choice. Internal standards should :
- not perturb or overlap the analyte spectrum,
- not interact with the sample, and
- be strong emitters, so that a small quantity added to samples and standards will produce signals of adequate intensity.
As these requirements, especially the last one, are difficult to be fulfilled, several other techniques have been used [17-19]. Amongst them, the most promising one is the use of an external standard [20, 21] that compensates not only excitation intensity fluctuations but other parameters, such as sample positioning changes.
In this work, normalization was achieved by dividing the spectrum by the intensity of the cyclohexane 802 cm-1 band and multiplying by 100.
The effect of normalization on the long-term stability of the calibration curves is shown in Table II.
| Table II. The effect of normalization against the 802 cm-1 cyclohexane band on the calibration curves. | ||||
| % Relative difference of the calibration curve slopesa | ||||
| Band intensity | Band area | |||
| Band, (cm-1) | Raw data |
Normalized data |
Raw Data |
Normalized data |
| 2951 | 52 | -3.7 | 73.9 | -6.3 |
| 1065 | 72 | 8.2 | 24.9 | -11 |
| 661 | 41 | -12 | 72.3 | -8.4 |
| 604 | 65 | 3.2 | 28.7 | -11 |
| 568 | 31 | -18 | 4.01 | -14 |
| a Calibration curves were acquired four days apart and under different excitation intensities (0.57 and 1.01 watt). | ||||
Differences in calibration curve slopes at 2951, 1065, 661 and 604 cm-1 bands were 41-72% and 3.2-12% before and after normalization, respectively. The effect of normalization was less pronounced for the 568 cm-1 band, as this is the weakest one. A second approach for normalization was also tried by using the 828 and 1205 cm-1 solvent bands as internal standard, but gross errors were obtained in formulation analysis and the linear range of the calibration curve was diminished.
By normalization, a Raman equivalent to the UV-Visible spectrophotometric absorption coefficient can be proposed. In our study, cyclohexane was found to be a convenient external standard, resulting in increased stability of calibration curves.
Quantitative analysis of fenthion formulations
The 2951, 1065, 661, 604 and 568 cm-1 bands were used for the quantitative analysis of fenthion, as the solvent (xylene, mixture of three isomers) and the surfactants do not interfere spectrally. Band area and intensity were calculated employing two point baseline correction, using the range
- 2943-2962 cm-1 for the 2951 cm-1 band,
- 1057-1076 cm-1 for the 1065 cm-1 band,
- 620-667 cm-1 for the 661 cm-1 band,
- 590-615 cm-1 for the 604 cm-1 band and
- 563-577 cm-1 for the 568 cm-1 band.
In Figure 2 one can see the spectra acquired during calibration, while calibration data are presented in Table III.
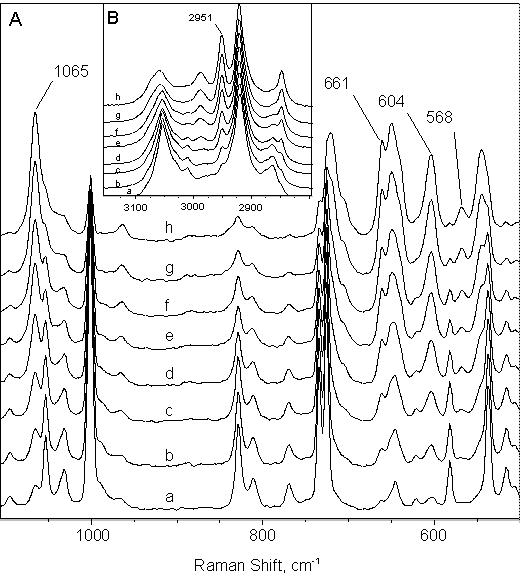
Figure 2. Raman spectra acquired during calibration: (a) 0.400, (b) 1.00, (c) 1.50, (d) 2.00, (e) 2.50, (f) 3.00, (g) 3.50 and (h) 4.00 M fenthion.
| Table III. Calibration data for fenthion at 1.01 watt excitation intensity. | ||||
| Band, (cm-1) |
Band intensity calibration curve |
ra | Band area calibration curve |
ra |
| 2951 | BI = (-1.6± 0.4) + (5.9±0.2) ´ C | 0.996 | BA = (-35± 7)+ (74±3) ´ C | 0.993 |
| 1065 | BI = (0.3± 0.2) + (2.94±0.08) ´ C | 0.997 | BA = (-8± 3) + (25±1) ´ C | 0.991 |
| 661 | BI = (-0.5± 0.1) + (1.09±0.05) ´ C | 0.992 | BA = (2± 4) + (72±2) ´ C | 0.993 |
| 604 | BI = (-0.8± 0.1) + (2.19±0.05) ´ C | 0.997 | BA = (-0.3± 1.3) + (28.7±0.6) ´ C | 0.998 |
| 568 | BI = (-0.25± 0.08) + (0.44±0.03) ´ C | 0.98 | BA = (-3± 1) + (4.0±0.4) ´ C | 0.97 |
| aCorrelation coefficient. | ||||
The calibration curves for band intensity and band area are presented in Figure 3. The linearity of the 2951, 1065, 661 and 604 cm-1 calibration curves is excellent. The correlation coefficients for band intensity and band area measurements were in the range of 0.992-0.997 and 0.991-0.998, respectively. The linear range of 0.4-4.36 M is convenient for quantitative analysis, as it permits formulation analysis without sample dilution and extensive manual handling of the toxic samples. The calibration curve for the 568 cm-1 band presents a linear range of 1-4.36 M. The detection limits for all bands except of the 568 cm-1 band, were in the range of 0.14-0.27 and 0.14-0.36 M for band intensity and band area measurements, respectively. The detection limits for the 568 cm-1 band were 0.54 and 0.75 M for band intensity and band area, respectively.
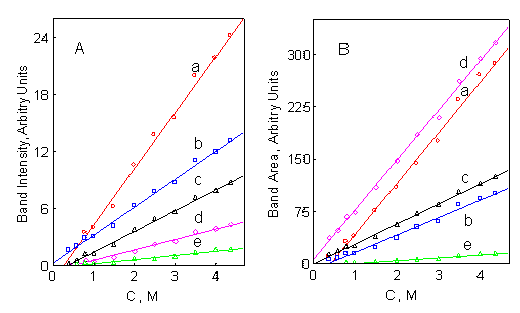
Figure 3.Fenthion calibration curves based on band intensity (A) and band area (B), acquired at (a) 2951, (b) 1065, (c) 604, (d) 661 and (e) 568 cm-1.
The only parameters that affect precision are the instrument parameters, as sample pre-treatment is not required and samples are homogeneous solutions. Amongst the factors affecting precision are differences in sample cells and positioning changes when exchanging samples [22]. The most dominant instrument parameter is laser instability within the time scale of the analysis [20]. The uncertainty in sample positioning and laser power is also reflected in detection limit values [23]. Precision estimation was achieved by changing the position of the sample cell between the acquisition of successive spectra. Precision for the 2951, 1065, 661 and 604 cm-1 bands, ranged from 0.57-4.7 and 0.4-6.8 %RSD, (n=4) for band intensity and band area measurement respectively, while for the 568 cm-1 band ranged from 0.92-8.0 and 1.8-8.8 %RSD, (n=4) respectively. The good reproducibility of FT-Raman spectrometers has been attributed to the large entrance aperture which permits focusing the laser into a relatively large sample volume [21].
The sensitivity of the analytical method increases by increasing the excitation intensity. In figure 4 one can see the effect of excitation intensity on the analytical signal. As shown, there is a linear relation between excitation intensity and the analytical signal. To compensate for the excitation intensity fluctuations a multiplication factor, such as the inverse intensity of the external standard can be used [20]. To achieve maximum sensitivity, a high excitation intensity should be used, provided that the sample is not thermally degraded and precision is not diminished.
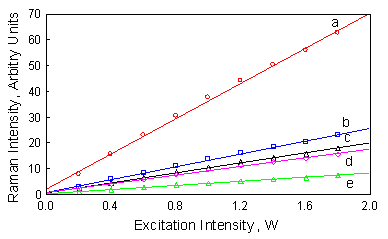
Figure 4. Effect of excitation intensity on band intensity measurements of the 3.50 M fenthion standard, acquired at (a) 2951, (b) 1065, (c) 604, (d) 661 and (e) 568 cm-1.
Band area measurements were used for the analysis of pesticide formulations, as interactions of sample components are more likely to influence the intensity of Raman bands by shifting, whereas the integrated intensities are less affected [22, 24]. Results of the analysis of the Lebaycid formulation by band area measurements are presented in table IV. Results obtained by the proposed method are equivalent to those acquired through the reference GC method [4], as shown by the t-test.
| Table IV. Analysis of Bayers’ Lebaycid formulationa by band area measurement. | |||
| Band,
(cm-1) |
Concentration found ± S.D. (M), (n=4) | t-valueb | |
| 2951 | 1.8 ± 0.1 | 0.251 | |
| 1065 | 1.82 ± 0.07 | 0.530 | |
| 661 | 1.9 ± 0.1 | 2.140 | |
| 604 | 1.75 ± 0.06 | 1.152 | |
| 568 | 1.8 ± 0.2 | 0.494 | |
| a Fenthion concentration 1.77 M ± 0.04, (n=3) asdetermined using the GC reference method.b ttheoritical=2.571 for 95% confidence level and five degrees of freedom. | |||
Conclusions
Quantitative analytical methods based on FT-Raman spectroscopy for organophosphorus pesticide formulation analysis [25-28] offer several advantages in comparison with existing methods:
Increased safety: Toxic samples are analysed “as received” avoiding sample pre-treatment.
Low analysis time: 30 s are sufficient for acquiring a spectrum region convenient for quantitative analysis.
Simplicity: Methods can be performed by minimally trained personnel.
Improved calibration stability: By using external standards.
Good agreement with time consuming official methods.
References
- C. D. S. Tomlin, The Pesticide Manual, (British Corp Protection Council, London, 1997) 11th ed., p. 531.
- Y. Hirano and T. Tamura, Anal. Chem., 36, 800 (1964).
- F. B. Ibrahim and J. C. Cavagnol, J. Agr. Food Chem., 14, 369 (1966).
- B. J. Gudzinowicz, Anal. Chem., 37, 439 (1965).
- D. MacDoucall, Anal. Methods Pestic. Plant Growth Regul. 2, 43 (1964).
- J. G. Grasselli and B.J. Bulkin, “Analytical Raman Spectroscopy”, (J. Wiley and Sons, New York, 1991).
- N. J. Szabo and J. D. Winefordner, Appl. Spectrosc., 51, 965 (1997).
- N. J. Szabo and J. D. Winefordner, Anal. Chem., 69, 2418 (1997).
- M. L. Nicholas, D. L. Powell, T. R. Williams and S. R. Huff, J. Assoc. Off. Anal. Chem., 59, 1071 (1976).
- A. M. Alak and T. Vo-Dinh, Anal. Chem., 59, 2149 (1987).
- P. A. Tanner and K. H. Leung, Appl. Spectrosc., 50, 565 (1996).
- H. Baranska, A. Labudzinska and J. Terpinski, Laser Raman Spectrometry Analytical Applications, (Ellis Horwood Ltd., Chichester, 1987), Chap. 4.
- M. L. Nicholas, D. L. Powell, T. R. Williams, R. Q. Thompson and N. H. Oliver, J. Assoc. Off. Anal. Chem., 59, 1266 (1976).
- J. C. Bowman and D. P. DeSalvo, Nicolet Application Note no. 9250, (1992).
- E. A. Cutmore and P. W. Skett, Spectrochim. Acta, 49A, 809 (1993).
- H. Sadeghi -Jorabchi, R. H. Wilson, P. S. Belton, J. D. Edwards-Webb and D. T. Coxon, Spectrochim. Acta, 47A, 1449 (1991).
- G. Jalsovszky, O. Egyed, S. Holly and B. Hegedus, Appl. Spectrosc., 49, 1142 (1995).
- X. Dou, Y. Yamaguchi, H. Yamamoto, H. Uenoyama and Y. Ozaki, Appl. Spectrosc, 50, 1301 (1996).
- X. Dou, Y. Yamaguchi, H. Yamamoto, S. Doi and Y. Ozaki, Vib. Spectrosc., 14, 199 (1997).
- C. J. Petty, P. J. Hendra and T. Jawhari, Spectrochim. Acta, 47A, 1189 (1991).
- T. Jawhari, P. J. Hendra, H. A. Willis and M. Judkins, Spectrochim. Acta,46A, 161 (1990).
- M. J. Smith and F. T. Walder, Nicolet Application Note no. 9145, (1991).
- D. R. Lombardi, C. Wang, B. Sun, A. W. Fountain III, T. J. Vickers, C. K. Mann, F. R. Reich, J. G. Douglas, B. A. Crawford and F. L. Kohlasch, Appl. Spectrosc., 48, 875 (1994).
- A. G. Miller and J. A. Macklin, Anal. Chem., 52, 807 (1980).
- S.G. Skoulika, C.A. Georgiou and M.G. Polissiou, Appl. Spectrosc. 53,1470 (1999).
- S.G. Skoulika, C.A. Georgiou and M.G. Polissiou, Talanta 51, 599 (2000).
- S.G. Skoulika and C.A. Georgiou, Appl. Spectrosc. 54, 747 (2000).
- http://www.aua.gr/georgiou/page20.html
REF: S. G. Skoulika, C. A. Georgiou and M. G. Polissiou Int. J. Vib. Spect., [www.irdg.org/ijvs] 4, 3, 4 (2000)