Infrared Spectra of Solids – the Mull Technique
Editor and Fabrice Birembaut*
*University of Southampton
Highfield, Southampton
SO17 1BJ, UK
Most analytical samples submitted to the infrared spectroscopist for identification (i.e. qualitative analysis) are solids. A variety of methods exist for their study including –
- The KBr disk method – described with examples in a piece by Geoff Dent in IJVS [volume 1, edition 1]
- Diamond ATR -again described in IJVS, this time by David Coombes [volume 2, edition 2]
- Reflection spectroscopy – we have talked about this, but perhaps we should look again
- Emission Spectroscopy – not sure we know much about this – perhaps we should know more AND
- The mull technique
When your dear Editor was a postgraduate a very long time ago, his supervisor – the late Dr Don Powell considered that this method was the best available so it will not be a surprise, we suspect, if we still think it’s worthwhile!
The basic principles
In order to record a transmission spectrum of a solid one needs to grind the particles to a size less than the wavelength of the transmitted wavelength and the particles should be immersed in a fluid with an index of refraction close to that of the solid micro-crystals.
If a crystal is suspended in a fluid with a perfectly matched index, the crystal will disappear – there will be no effective optical interface and light will transmit freely – no scattering will occur. If the index match is close but not perfect somereflection (i.e.scatter) will occur. The greater the mis-match – the more intense the scatter.
Quite obviously, each sample will have its own index of refraction. Further, the index is orientation dependent AND the index changes as one goes through an absorption band. Thus, it is impossible to find a fluid in which to immerse a crystallite to acquire perfect index matching. On the other hand, intelligent choice could well result in a ‘close match’ and hence a low level of scatter.
If crystallites are suspended in air and are much smaller than the wavelength of the radiation passing around them, they cease to exist optically as much. If the size is very much less than the wavelength they behave almost as a liquid, if the size is a little less than the wavelength both scatter and some transmission will occur. If these particles are now immersed in fluid roughly matching the indices of refraction the scatter can be reduced and hence we have a true transmission spectrum. Diagrammatically we have –
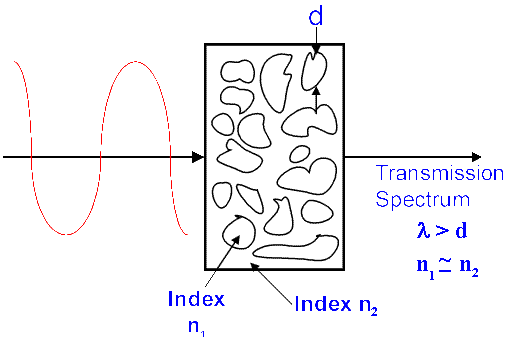
Figure 1. Transmission spectra of solids.
This experimental situation can conveniently be achieved in two ways:
- The KBr disk technique – where the suspension fluid is crystalline potassium bromide compressed so intensely that it behaves like a fluid and flows around and in optical contact with the particles and
- The mull method. Here, the material is ground up and then suspended in an appropriate organic liquid as a paste. The liquid has to satisfy several requirements – it must be free flowing, have a very simple and reproducible infrared spectrum and be readily available, cheap and stable.
The KBr technique is described in an article in Volume 1, Edition 1 of IJVS. In the mull technique the fluid of original choice was a particular commercial grade of liquid paraffin renowned for its laxative properties – Nujol, now immortalised as the “Nujol Mull”. I understand Nujol is no longer available, so we spectroscopic folk must resort to good quality liquid paraffin from their local pharmacists. This too is becoming hard to come by, because pharmacists are now instructed to be suspicious of customers purchasing same if there is any risk that they might be anorexic. So – sorry – slim, young lady purchasers must ask their overweight male colleagues † to purchase the stuff!
† Neither of your esteemed authors are sylph like – hence they have not encountered this problem!
The transmission spectrum of a thin film of liquid paraffin is shown below in Figure 2*.
*Editor’s Note – Don Powell would have told me to go to ‘Boots the Chemist’ to buy a new bottle of Nujol if he had seen this – it’s dreadful. Such is progress!!
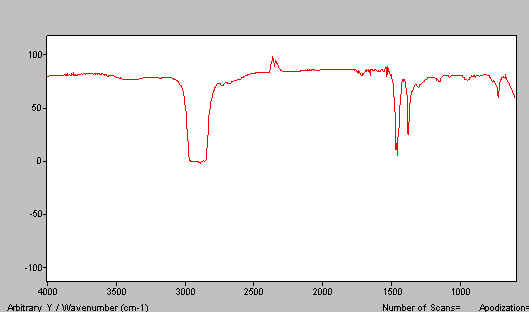
Figure 2. The IR spectrum of liquid paraffin.
Quite obviously, if you disperse a solid in this medium you will always see the paraffin bands. These arise, of course, from u, d & t CH motions near 2900, 1400 and 720cm-1respectively. Of these, the stretches and deformations are the most persistent. We need an alternative dispersing agent (mulling fluid). The classical one and I suspect still the best, is hexachlorobutadiene (HB). This material is very oily, relatively non-volatile and has a usefully high index of refraction. HB has, of course, a very considerable and intense absorption spectrum in the mid i.r. but since it contains only C-C and C-Cl bonds it is devoid of bands above 1700cm-1and also in the region 1500-1200cm-1. Thus, a combination of paraffin and an HB mull reveals the whole spectrum. Unfortunately, hexachlorobutadiene is toxic hence alternatives are recommended of which fluoro-carbon oils are popular. However, assuming you don’t take a bath in the stuff and/or breath in deeply of its rather pleasantly scented vapour, we suspect HB is unlikely to bring any of you ‘out in spots’ and is definitely an excellent mulling medium#. The spectrum of HB is shown in Figure 3.
# Caution – in some countries you MUST use safer alternatives, if they are available. You must therefore follow local legal requirements.
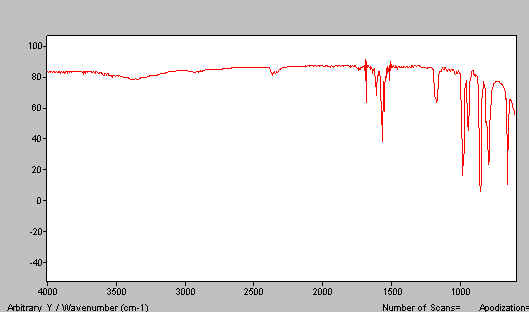
Figure 3. IR spectrum of hexachlorobutadiene.
Combining Figures 2 and 3, you can see the point – two mulls cover the whole useful spectrum.
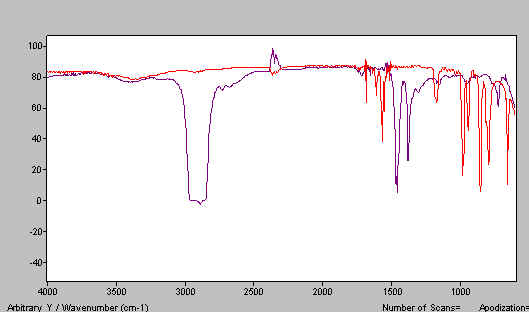
Figure 4. The two spectra Nujol and HB, one above the other.
You will need a small mortar and pestle made of agate. These are available from spectroscopic accessory makers. You also need a stainless-steel semi-micro spatula, two dropping bottles containing liquid paraffin and the alternative mulling agent of your choice (HB, Fluoro-carbon oil…) and plenty of tissues.
Making Mulls
The mull you make will be squeezed between two KBr flats so you will have to lay your hands on a pair in good condition. If they need polishing – see your Editor’s recent piece on the subject[volume 4, edition 1].
The way you prepare a mull depends to some extent on whether your sample is organic or inorganic. Relatively soft materials – and these include the vast majority of organic materials, co-ordination compounds and some mineral type materials. Others are very hard e.g. ionic crystalline and mineral substances. Further, the index of refraction of MOST but not ALL organics will be around 1.4-1.6. Many inorganics have higher values so we have an unfortunate pairing of properties:
| Organics/Co-ordination compounds | SOFT | & MODERATE INDICES |
| Inorganics especially crystalline materials | HARD | & HIGHER INDICES |
Thus, it is easy to make mulls from most organics and quite a bit harder for the hard materials.
| The stages involved are: |
|
|
|
|
|
|
|
|
Let’s start with an organic. We chose at random benzophenone.
A little less than 5mg of the material was transferred to the agate mortar. Dry grinding in this casedoes not work. The material simply forms a pad on the lower surface of the mortar. If this happens clean out the mortar, add new material and DON’T dry grind.
Add 1 drop of mulling fluid (a drop is around 1/20ml) and gently grind the mixture. As you do so, the lightly ground material tends to form a ring around the outer surface of the pestle so you should touch this off as you grind to minimise the risk that some of the material will not be really well ground. Grind for about 30 seconds.
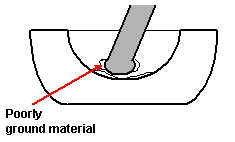
Figure 5
Scrape the mull into the centre of mortar and test its viscosity. The viscosity should be similar to that of tomato ketchup. If too viscous add a small further drop of mulling fluid and regrind for 15-20 seconds to mix.
Again scrape the mull into the centre of the mortar and transfer to one of the KBr flats. Do not bother to try and transfer everything – you will only need about half. Add the second flat and then gently rotate and press downwards. Gently swirl the top plate over the bottom plate. DO NOT rotate. The motion is the same as the one used in polishing, but on a much smaller scale.
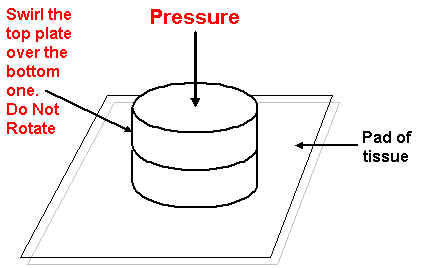
Figure 6
The mull will spread out into a roughly round layer. DO NOT press too hard. DO NOT slide the top flat too violently, be gentle or you will scratch the KBr flats.
Put the flats as a sandwich into the spectrometer and run the spectrum. If you have ground the mull properly and the ratio of benzophenone to liquid paraffin is about right, you should achieve a spectrum like this.
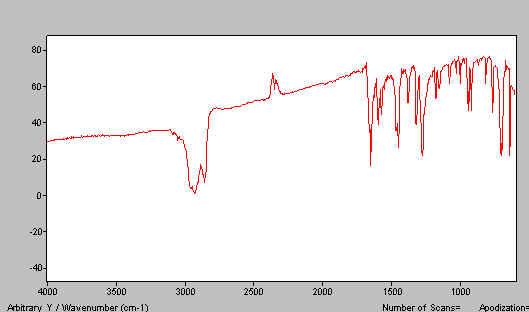
Figure 7. Benzophenone spectrum – paraffin mull
You are unlikely to achieve this result first time. There are several problems you might find:
A. Sample thickness too large OR you have squeezed too hard and the thickness is too thin. See Figure 8.
B. You have too much liquid paraffin and hence too little benzophenone. See Figure 9.
We suggest you now make a mull and try to match Figure 7. Once you have done it a couple of times you will “get the feel” and have no problem in the future.
C. If you don’t grind the material enough, you are unlikely to have a problem if the sample is soft. As we shall see below, this can be a problem with inorganics.
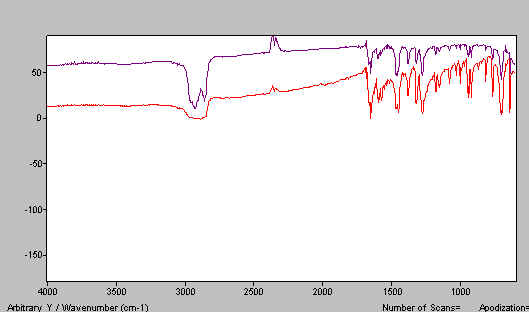
Figure 8. Benzophenone mull spectra – mull layer too thin.
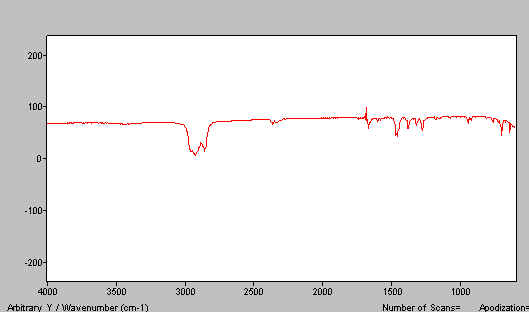
Figure 9. Benzophenone mull spectra – too much paraffin.
Now clean the mortar and pestle with tissue. If you are in any doubt use some solvent – say acetone on the tissue. Clean the KBr flats. Use of some solvent here is strongly recommended. If the KBr flat is difficult to clean or has become scratched give it a polish before proceeding.
Now make a second mull using the alternative mulling agent. We used HB and our effort is shown in Figure 10. In Figure 11 we combine the two mull spectra i.e. Figures 7 and 10 to produce the complete spectrum.
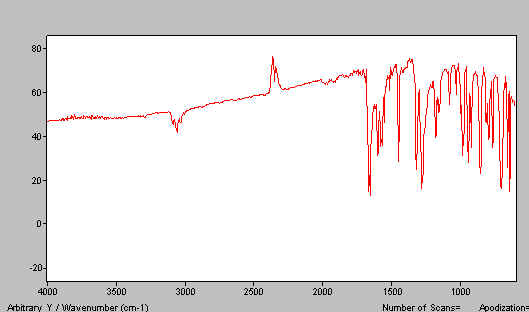
Figure 10. Benzophenone mull spectra – HB mull.
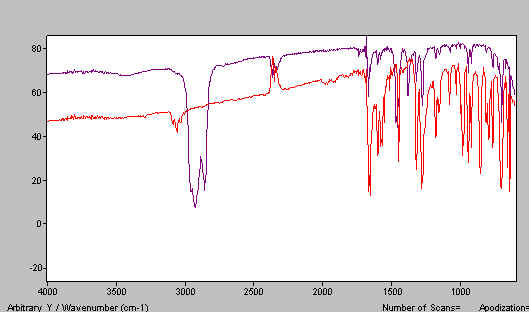
Figure 11. Combined mull spectra on benzophenone.
Organic samples rich in OH or NH groups are subject to high levels of hydrogen bonding. Such samples tend to give rather diffuse infrared spectra. H bonding broadens the bands and they can frequently overlap making the spectrum appear to be poorly resolved. There is no doubt that the quality of the sample preparation has a role here.
In Figure 12 we show a paraffin mull of lactose containing too much poorly ground lactose. Do the job properly and the spectrum improves – see Figure 13. A good HB mull appears as the next figure (Figure 14).
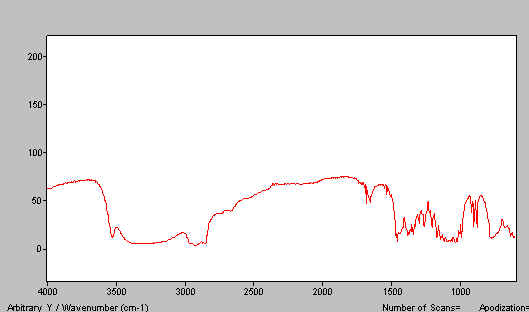
Figure 12. Lactose mull spectrum – paraffin mull – a poor example.
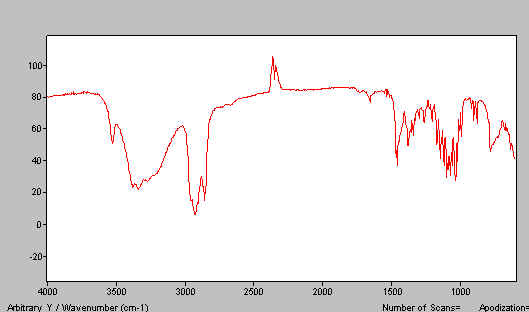
Figure 13.
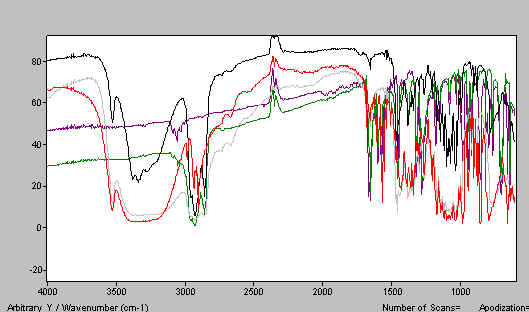
Figure 14. Lactose mull spectrum – paraffin mull.
Now Inorganics
Proceed exactly as you did for benzopherone but emphasis must be put on grinding. At each stage be really careful that ALL the mull is ground finely. So, be very careful that the material that collects around the edge of the pestle is returned to the mortar several times.
Quite arbitrarily we chose potassium di-chromate. Remember, the problems here are that the material is much harder than is typical of organics AND the index of refraction of many (but not all) inorganics can be quite high.
If you do the job properly, you should achieve a good mull and a spectrum like that shown below in Figure 15. If you don’t grind properly and/or have too much solid, spectra like that shown in Figure 16 is typical.
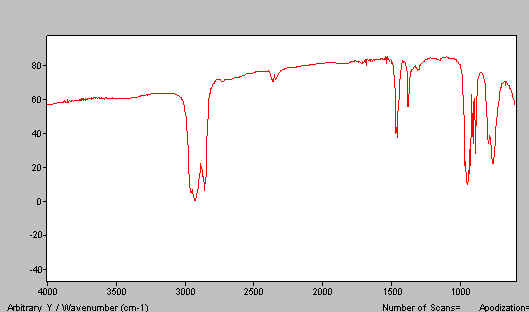
Figure 15.
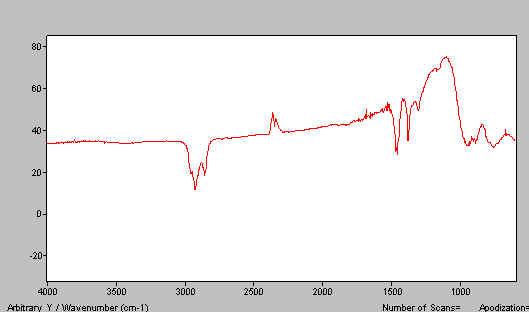
Figure 16. K2Cr2O7 paraffin mull spectra.
Let us say your spectrum is too thick. The temptation is to press the flats closer together and to move the top flat around to smear out the sample. This works but is very likely to cause scratching of the KBr surfaces. A trick that sometimes works is to separate the flats, add a tiny drop of paraffin to one flat and rejoin. Gentle movement of one flat with respect to the other causes mixing, thinning and probably some grinding. The mull in Figure 17 was given this treatment and produced much better results. The index of refraction of HB is high, making it much better as a mulling agent than paraffin for inorganics. Very little effort was required to produce Figure 17.
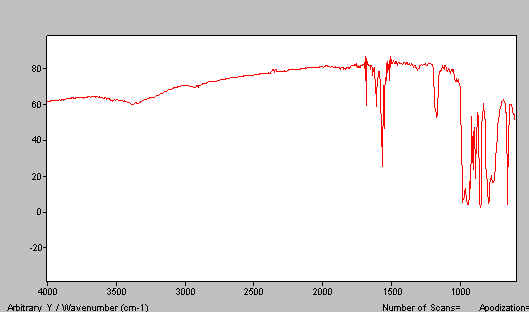
Figure 17. HB mull spectrum of K2Cr2O7
So- it is easy and quick to make a mull and run it’s spectrum. We estimate making the mull and putting it on the flats takes about as long as recording the spectrum. That is, the mulling method is certainly quicker than the KBr disk method. On the other hand, the mull always contains the bands due to the mulling agent. So – is it worthwhile particularly now that diamond ATR is so widely available?
The answer is yes because, there is another excellent reason for using the mull method – reduced risk of damage to the sample.
Inorganic chemists are well aware that exposing mixtures of their compounds with potassium bromide at extremely high pressures is seriously risking the possibility of reactions. Ionic compounds could re-equilibrate. Reactive chlorine or iodine atoms could well interchange with the massive excess of bromide. But – there is yet another hazard – polymorphism.
Many materials exist in polymorphs i.e. they adopt more than one crystal structure. These structures inter-convert but many are meta-stable. Thus, although not the most stable form at room temperature, they either only slowly convert or they don’t convert at all. Polymorphism is of vital interest in several areas of the fine chemical industry – dyes, additives and pharmaceuticals. The main reason is that the rate at which substances dissolve in solvents is governed by their polymorphism. Take the wrong polymorph of your pharmaceutical and it may do nothing!
Unfortunately for analysts, the application of extreme pressure WILL re-equilibrate polymorphs so the use of the KBr disk or diamond ATR methods is risky. Both MAY be OK but both ARE RISKY. The mull procedure is far less likely to cause re-equilibration.
Holding mulls in your spectrometer
Most of the accessory manufacturers offer ‘mull holder’ consisting really of a simple clamp to hold the sandwich of KBr flats in the beam. In our opinion they are clumsy, relatively awkward to use and if used incorrectly the sandwich can become unstuck. There is a simple alternative – a simple support for the sandwich applying no clamping force, a solution you can make yourself. We show below two designs.
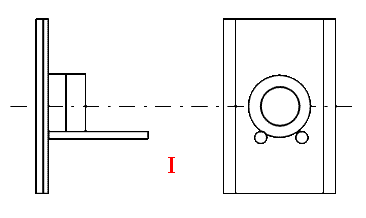
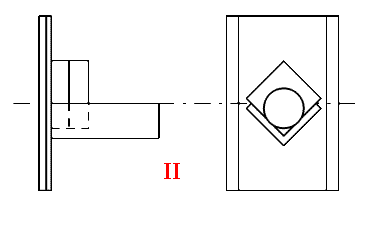
Both are shown on 2”x3” Industry Standard ‘cards’. One simply consists of two appropriately cited stainless steel rods upon which round or square KBr flats can sit. We use 1/8” diameter rod (3mm is fine). The second uses a short section of aluminium ‘angle’ to support the sandwich. We recommend 3/4” x 3/4” x 1/8” material.
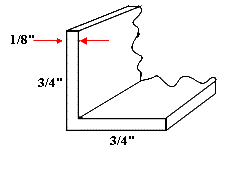 This material is widely available. Cut a length of about 15mm – clean up the sawn ends and stick it on the card with epoxy.
This material is widely available. Cut a length of about 15mm – clean up the sawn ends and stick it on the card with epoxy.
A reasonable competent “do-it-yourselfer” should have no problem. Oh yes – ‘industry cards’. Most infrared labs have the odd card usually made out of stainless steel lying about.
Good luck
Click here to view spectra for this article
REF: P.J. Hendra & F. Birembaut, Int. J. Vib. Spect., [www.irdg.org/ijvs] 4, 3, 3 (2000)