pH Dependent Surface Enhanced Raman Scattering of Molecules Adsorbed on Silver Hydrosol
Joydeep Chowdhury+, Manash Ghosh+,
Kana. M. MukherjeeÑ and T. N. Misra+*
+ Department of Spectroscopy,
Indian Association for the Cultivation of Science,
Jadavpur,
Calcutta : 700 032, India.
Ñ Department of Physics,
Malda College,
Malda. India
* e-mail of the corresponding author : sptnm [at] mahendra.iacs.res.in
Abstract
The pH dependence of the Surface Enhanced Raman spectra (SERS) of pyridine-4-carbaldehyde, hypoxanthine and 8-hydroxyquinoline is reported. Such dependence of SER spectra has been explained in terms of the state of protonation and hydration of pyridine-4-carbaldehyde molecule while for hypoxanthine, it is considered to be due to the change in the molecular orientation and keto-enol tautomerism. The variation of SER spectra with pH for hydroxyquinoline is attributed to the adsorption of the molecules on the bare and the chlorinated silver surfaces whose surface density depends on the sol pH.
Introduction
The discovery and the development of Surface Enhanced Raman Scattering (SERS) has opened up wide research fields both in the physics and chemistry of interfaces [1-3]. It has been realised for the first time that there exists a high resolution technique for studying trace concentration of materials adsorbed on a restricted range of metal surfaces. The enhancement of surface Raman scattering per molecule ranges from 103 to 106 for the first adsorbed monolayer. This enormous enhancement factor enables one to determine detailed structural information about the adsorbed molecules even in sub-monolayer coverage. SERS study has been extended to the identification of complex molecules at trace levels and the investigation of the mechanisms and kinetics of chemical or biological reactions at the interfacial regions.Introduction
Raman scattering arises from the induced dipole moment (P) produced by the interaction of electromagnetic field of light (E) with the molecular polarisability a mathematically connected by
P = a E ………………………………….(1)
where P and E are vectors and a is a tensor of rank 2. So the origin of the tremendous enhancement in Raman scattering is obviously due to perturbation in either or both of E and a . Based on this fundamental concept, it is now unanimously accepted that two simultaneously operative mechanisms, a long range ” classical electromagnetic effect” [EM] attributed to the increase in the local field of the adsorbate due to the excitation of the plasmons on the metal surface [4] and a short range “chemical effect” [CHEM] arising due to perturbation of the adsorbate polarisability (a ) resulting from the electronic interaction between the molecule and the metal surface [5-8] are responsible together for the large enhancement of the Raman scattering.
In SERS, when colloidal metal particle surfaces are used as the adsorbing sites, the pH of the sol can be controlled. It is observed that as the sol pH is changed, many molecules at the silver-water interface undergo transformation such as chemical reaction, cis-trans isomerization, keto-enol tautomerism or orientational change of adsorbed molecules due to change of adsorption sites. Here we report the pH dependence of SER spectra of pyridine-4-carbaldehyde (4-PCA), hypoxanthine (HX) and 8-hydroxyquinoline (HQ) adsorbed on colloidal silver particles. Their SER spectra show that the variation of the SER spectra at different pH is attributed to the degree of hydration and protonation of 4-PCA at the silver-water interface, the change in molecular orientation and the keto-enol tautomerism for HX and to the adsorption of the molecules on bare and chlorinated silver surface for HQ.
Experimental Methods
Chemicals and Procedure
4-PCA, HX and HQ were purchased from the Aldrich Chemical Co, U.S.A. 4-PCA was purified by vacuum distillation under reduced pressure in a nitrogen atmosphere whereas HX and HQ by vacuum sublimation prior to use. The 4-PCA molecule is soluble in water while HX and HQ are readily soluble in dilute mineral acids. The molecules were adsorbed on the metal colloids to record their SER spectra. Dilute HCl and NaOH solutions were used to adjust the pH value of the mixture which was measured by a pH meter (Systronics m pH system, Model 361)
Colloid preparation
Although SERS has been observed on many types of substrates, the small metal colloidal particles have been proved to be much more advantageous over solid surfaces because of experimental ease and simplicity of sol preparation. Chemical reduction of metal salts is one of the most frequently used techniques for preparing colloidal metals.
Silver Colloids
Silver colloids are prepared according to two different recipes. In one, the silver hydrosol is prepared by reducing AgNO3 with sodium borohydride as a consequence of adding 100 ml of AgNO3 (6mM) solution dropwise to 300 ml solution of NaBH4(1.2 mM) with slow stirring. Both solutions are chilled by ice-cold water prior to mixing [9]. The prepared Ag colloidal solution has a single absorption maximum at 392 nm and consists of roughly spherical Ag particles of diameter in the range 1-50 nm. The colloid thus prepared is yellow in colour and is stable at room temperature for several weeks.
In the alternative method, sodium citrate is used as the reducing agent [10]. In this method 50 ml of 1mM AgNO3 solution is heated near it’s boiling point and then 1 ml of 1% sodium citrate is added to the heated AgNO3 solution. The solution is then stirred and finally boiled for 5 minutes. The sol thus prepared is greenish yellow in colour and show an absorption maximum at 420 nm.
Copper and Gold Colloids
Beside silver colloids, copper and gold ones are also used extensively for SERS studies. Cu colloid is prepared by adding 1 ml of aqueous solution of copper (II) sulphate (1 x 10-2 M) in 25 ml of trisodium citrate solution (5.6 x 10-3 M) and then adding 12 ml of a freshly prepared solution of sodium tetrahydroborate (2 x 10-2 M) in sodium hydroxide (2 x 10-2 M) [11]. The resulting brown colloid is aged for at least 1.5 hours before being employed in SERS measurements. The colloid becomes dark red and shows an absorption maximum at 560 nm.
There are two different methods generally available for gold colloid preparation [12]. In one, the Au colloid is prepared by the partial reduction of KAu(CN)2 (20 ml, 3mM) with ice cold aqueous NaBH4 (40 ml, 10mM) purged with N2. Immediately after mixing, the solution is heated to about 800 C for 30 minutes and is then cooled to room temperature. In the second method, the colloid is prepared by the reduction of KAuCl4(10 ml, 3mM) with ice-cold aqueous NaBH4 (10 ml ,10mM) purged with N2. Upon warming to room temperature, this mixture acquires a red appearance.
Sutherland et.al [13] used a new methodology for Gold colloid preparation. In this method 0.1 ml of HAuCl4 solution ( 1% w/v) was added to 40 ml of triply distilled water and then 1 ml of trisodium citrate solution (1% w/v) was added drop by drop while stirring and the resulting mixture was boiled for 5 minutes. The solution obtained was raspberry-red, showing an absorption maximum at 520 nm. This sol was used by Camafeita et.al for SER measurements [14].
Recently laser ablation of metals has been successfully used to prepare SERS-active colloids [15,16]. The major advantage of preparing SERS-active substrates by laser ablation is that the surface of such colloids is free of organic or ionic contaminants. This has been extended more recently for the preparation of silver island films which have been used to study the SER spectrum of Cresyl Violet and Crystal Violet.[17]
We have used silver hydrosol prepared by the Creighton procedure [9] for our experiments. All required solutions were prepared with distilled and deionized water from Milli-Q-plus system of M/S Millipore Corporation, U.S.A.
Recording of Raman spectra
Raman spectra were recorded on a Spex double monochromator (model 1403) fitted with holographic gratings of 1800 grooves/mm and a cooled photomultiplier tube (Model No. R928/115) from Hamamatsu Photonics of Japan. The water soluble samples were added to the silver hydrosol in a measured amount to have the desired solute concentration in the sol. For water insoluble samples, their solution in a suitable organic solvent was added to the sol and dispersed uniformly (in some cases by sonication) until some molecules were adsorbed on the colloidal particles. The sample, in a quartz cell was placed in the sample chamber which facilitated optimum illumination of the sample and collection of scattered radiation. The sample was excited through the bottom face of the cell with 514.5 nm radiation from a Spectra Physics Ar+ ion laser (model 2020-05) at a power of 200 mW for the NRS in solution and 100-250 mW (depending upon the sample) for SERS measurements. Raman scattered radiation was collected at a right angle to the excitation. The operation of the monochromator, the photon counter and the data acquisition and analysis unit were controlled by a Spex Datamate 1B. The inbuilt software facilitated background subtraction and data accumulation of large numbers of spectra. The acquisition time by the spectral element was 0.5 s. The scattered light was focused onto the entrance slit of bandpass 4 cm-1. All spectral measurements were made at least three times to ensure reproducibility.
Results and Discussions
pH dependent SER spectra of 4-PCA:
We characterised the conformation and structure of 4-PCA adsorbed on silver surfaces as a function of solution pH with the aid of SER spectra. In aqueous solution many aldehydes exist in two forms, the free aldehyde and the hydrate or gem-thiol related by a hydration equilibrium constant Kh.
![]()
Kh= ![]() , in large excess of water, where in our case R denotes the pyridyl ring. Also, many substituted pyridines, in the presence of H+ ion in the environment, undergo protonation at the ring nitrogen.
, in large excess of water, where in our case R denotes the pyridyl ring. Also, many substituted pyridines, in the presence of H+ ion in the environment, undergo protonation at the ring nitrogen.
We show that the presence of the metal surface modifies the extent of hydration and protonation significantly and that if the pH of the solution is varied, kh reaches an optimum value for which the adsorbate molecule is completely hydrated showing a SER spectrum identical with that of 4-pyridyl-methanol (4-PM). Hydration of the aldehyde group is an addition reaction caused by the nucleophilic attack [18] of a water molecule on the positively charged carbon atom of the polarised carbonyl group (Figure 1). An electron rich nucleophilic reagents, such as Lewis bases, when involved in a reaction donates electrons to its positively charged counterpart. When 4-PCA molecule is adsorbed onto the metal surface, this positivity of carbon atom increases. As a result, nucleophilic attack by water also increases and hydration of the aldehyde group occurs.
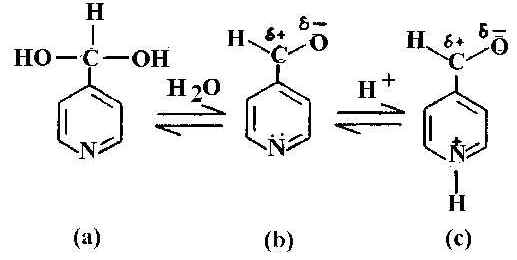
Figure 1. Schematic representation of pyridine-4-carbaldehyde: a) hydrated b) normal and c) protonated form.
It is interesting to examine the acid-base equilibrium of 4-PCA. In acidic media, owing to the availability of H+ ion, the pyridine nitrogen becomes protonated. Hydration and protonation of 4-PCA is shown in Figure 1. In NRS of this molecule, the carbonyl band at about 1711 cm-1 is prominent in the neat liquid and in aqueous solution. This band is not observed in SERS or in acidic or strong basic medium. Another notable feature is the behaviour of the pyridyl ring mode at 1600 cm-1 seen in the neat liquid and also in aqueous solution. In the NRS of the acidic solution this ring mode is absent and is replaced by a new band at about 1650 cm-1 This is the protonated ring mode. The protonation of the pyridyl ring is shown in Figure 1c. In the NRS of the basic solution such a pyridyl ring mode at 1600 cm-1 is present, as is a new band around 1360 cm-1. The appearance of this band, assigned as asymmetric C-O stretching [19] mode, instead of nC=O (C=O is suffix to n) mode, indicates that the adsorbate molecules do not exist in the aldehyde form. The band at 1365 cm-1 in the SER spectrum (Figure 2) is totally absent in neutral solution and also in neat liquid. This is assigned to the n [C-OH] mode, which arises on conversion of the aldehyde to its hydrate. In the SER spectrum at near neutral pH ( Figure 2c) both the pyridyl ring modes and the protonated ring modes are present.
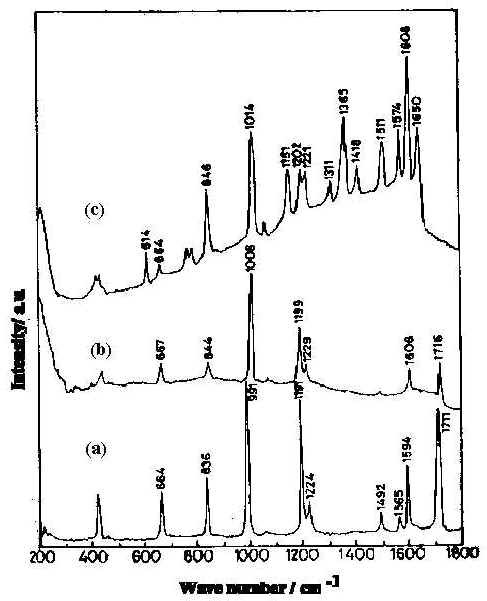
Figure 2. Normal Raman spectra of pyridine-4-carbaldehyde :
a) pure liquid b) 0.01 M aqueous solution at pH 6.5
and c) 8x 10-3 M in silver sol at pH 6.5.
[laser power 0.2 W , l = 514.5 nm]
Formation of the hydrate is evidenced by the appearance of the C-OH in-plane bending mode [20] at 1420 cm-1 in the SER spectrum (Figure 2c). The hydration of the surface species is responsible for the disappearance of the carbonyl band. This has previously been reported for metal electrodes [21].
The SER spectra of the 4-PCA molecule in the spectral range 1600-1750 cm-1 in the pH range 5-8 ( Figure 3) is interesting as both the pyridyl ring and the protonated ring modes are present and their relative concentration on the silver surface at varied pH can be estimated [22]. At the alkaline pH of 7.75 only the normal ring mode is observed. The considerable decrease in intensity of the normal ring mode along with the detoriation of the general spectral quality at this pH is due to enhanced background arising from the decreased optical quality of the sol. As the pH of the sol decreases, protonation increases and at pH 5.67, the pyridyl ring mode is intense and the protonated ring mode is weak. At pH 5.19, the pyridyl ring mode is of comparable intensity with the protonated ring one. At the lower pH of 5.14, the SER intensity of the normal ring mode decreases whereas the band due to the protonated species is greatly enhanced. This implies that as the sol becomes more acidic, the relative intensity of the band at 1650 cm-1 increases indicating that more pyridyl rings are protonated. Such protonation occurs up to pH 5.98; above which the pyridyl ring is not protonated. Thus by studying the SER spectra of 4-PCA molecule at various pH values of the sol, the extent of protonation at the silver-water interface can be monitored. The relative population of protonated and neutral hydrate on the silver surface can be estimated by calculating the intensity ratio of I1650 / ( I1600 + I1650 ), assuming this to be equal to the fraction of pyridine species on the surface that is protonated and the scattering cross section to be the same. Using this relationship the estimated fraction of protonated hydrate is 60 % in the pH region 5<pH<6, more than 60 % at pH<5 and for pH value >6, only normal hydrate is present.
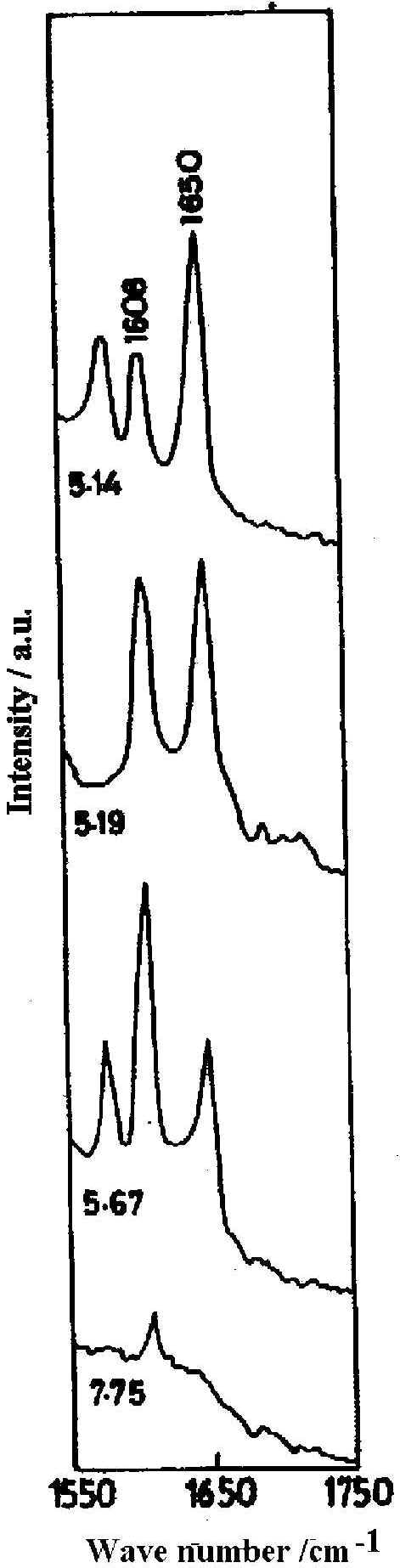
Figure 3. SER spectra of 4x 10-2 M pyridine-4-carbaldehyde at different pH
[laser power 0.2 W, l = 514.5 nm]
The fraction of aldehyde that is completely hydrated, may be rapidly and efficiently converted into the alcohol, 4-pyridyl methanol [23] as is reflected in the resemblance of the SER spectrum of 4-PCA and 4-PCM (Fig. 4) at pH 6.45. This result indicates that a reduction process is operative in SERS at near normal pH. Indeed a change in surface potential of silver with the change of pH may facilitate such reduction process. Thus SER spectroscopy allows us to identify surface species which may often be quite different from that found in the bulk solution.
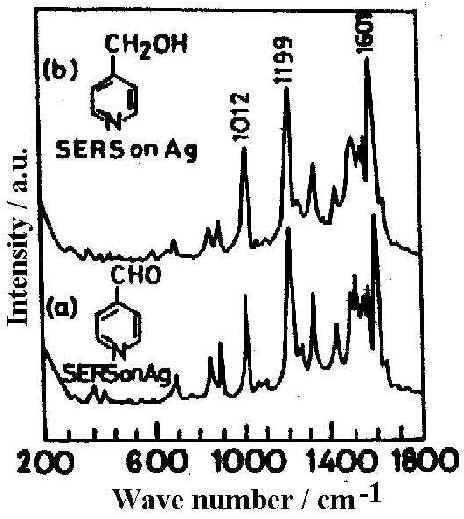
Figure 4. SER spectra on Ag surface at pH 6.45 from different solutions: a) pyridine-4-carbaldehyde, b) 4-pyridylmethanol
[laser power 0.2 W, l = 514.5 nm]
pH dependent SER spectra of HX
The HX molecule is known to exist in two tautomeric forms, namely the keto and enol tautomes. These are shown in Figure 5 (a &b). Normal Raman spectra of the HX molecule in the solid state and in dilute HCl solution(0.01M) have been recorded. Compared to the solid state spectrum, few bands appear in the solution spectrum. The SERS spectrum, however, contains a large number of bands whose intensity and position change with the pH of the solution.
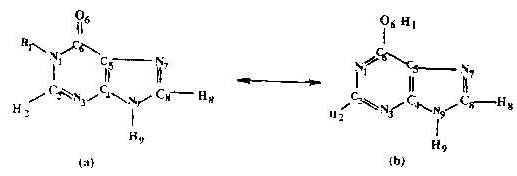
Figure 5. Structure of hypoxanthine molecule:
(a) Keto-form ; (b) Enol-form.
Figure 6 shows the SER spectra of the HX molecule (1 x 10-3 M) at different solution pH.s. Interestingly, the ring breathing mode in the SERS spectra at 725 cm-1 which is very strong in the range of acidic and neutral solutions is blue shifted to 746 cm-1 at alkaline pH.s. Such changes in the position of the ring breathing mode, which is quite common for this type of biomolecules, may either be due to the change in the adsorption geometry of the molecule on the surface or due to a change in the structure of the adsorbed molecule resulting from protonation of the N7 atom or due to keto-to-enol tautomerization [24]. In fact,the SERS spectra reveal that in the surface adsorbed state this molecule exists in the keto or the enol form depending on the pH of the sol.
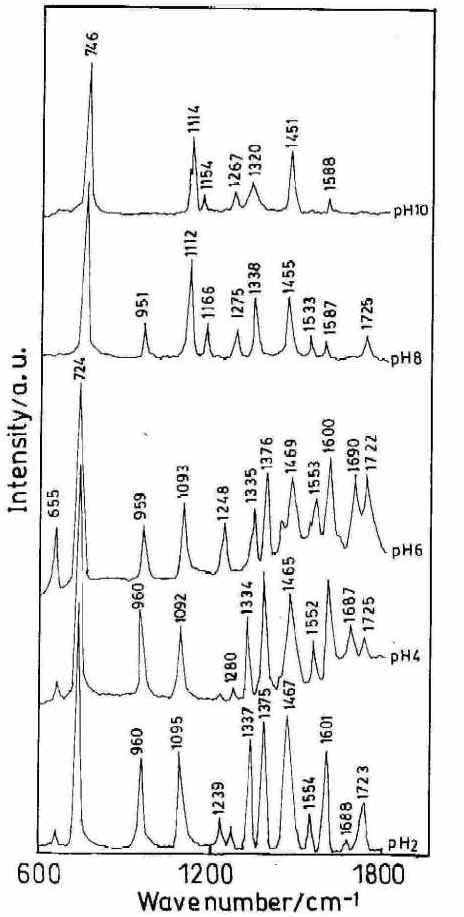
Figure 6. SERS spectra of hypoxanthine (high frequency range) at different pH. [laser power 0.25 W, l = 514.5 nm]
Interestingly, the carbonyl stretching mode assigned at 1678 cm-1 and the carbonyl in–plane bending mode [25] at 655 cm-1 are both very weak in the SER spectra at pH 2 and 4 and increase towards neutral pH (Figure 6) which implies that the HX molecule exists as the keto– N 9 H 9 form in the surface adsorbed state only at acidic and normal pH.s. Carbonyl stretching and bending modes are absent in alkaline pH where a new band at 1166 cm-1 appears. This mode is assigned as the C6– O6 H1 stretching mode. [25,26].
These results infer that the HX molecule exists in the (enol- O6 H1, N9 H9) form in the surface adsorbed state in alkaline pH. The presence of the N- H stretching mode [27] at 3237 cm-1 and also the N- H bending mode [28] at 1600 cm-1 over the whole range of pH.s can be rationalised as the N9 H9 group remains unperturbed in both the tautomers. This further implies that H9 atom does not form a C6- O6 Hydrogen bond. The H1 atom being nearest to C=O bond, is more likely to be shifted to form the C6-O6 H1 bond.
Precise determination of the adsorption site of the molecule on the Ag surface is fraught with problems. We see from the chemical structure of the HX molecule (Figure 5a and 5b) that it can exist in keto and enol tautomeric forms. In the keto form the molecule has two nitrogen atoms N3 and N7 capable of forming bonds with the silver surface and any one or both of them can be involved in chemisorption to silver, while in the enol form there are three nitrogen atoms N1, N3 or N7 and one oxygen atom appropriate for chemisorption which make the problem more complicated. The best way of treating this problem is to enumerate the negative charge density on each of these probable active sites. The higher the negative charge density on the atom, the higher is the probability of it to acting as an adsorptive site for the silver substrate [29]. In order to have a proper understanding of the ground state property of the molecule, a more precise geometry optimization was obtained using MOPAC’s NLLSQ gradient minimization routine with the AM1 Hamiltonian, [30-32] whereon energy of less than 0.12kcal/mol was used as the optimization criterion. With this optimized geometry , the negative charge density on the probable atomic sites of both the keto and the enol forms of the HX molecule has been calculated.
Results show that in the keto form the negative charge density on N7 and N3 atoms are -0.0614 and -0.1786 respectively while in the enol form it is -0.1757, -0.3978, -0.2415 and -0.2263 for the N7, O6, N1 and N3 atoms respectively. Negative charge density is higher on N3 in the keto form whereas in the enol form the negative charge density is significant on all the four atoms. However the metal-adsorbate interaction becomes more evident from the SERS spectra at different pH (Figure 7) which shows significant changes as the pH of the sol is varied from acidic to alkaline. SERS spectra of the HX molecule at acidic pH (pH 2) shows a strong band at 238 cm-1 which is assigned to Ag-N3 stretching mode. This band intensity gradually decreases with the increase in pH. Another band at 220 cm-1 appears with the increase of pH value (pH 6) and becomes gradually stronger and shifts towards the blue as the pH is increased. At pH 10 the band shifts to 214cm -1. This band is assigned to the n (Ag-O) mode. However, the intensity of this SERS band changes significantly with pH value and may indicate this band arises from interaction between the molecule and the metal surface through the formation of both Ag-O and Ag-N1 bonds. Our theoretical calculations show that though the negative charge density is higher on O6 and on N1 atoms, it is significant also on N3 and N7. In alkaline pH it, therefore, seems likely that all the four, O6, N1, N3 and N7, atoms are the active sites for the adsorption process. Hence we require two modes of orientation of the adsorbate on the substrate under acidic and alkaline conditions respectively.
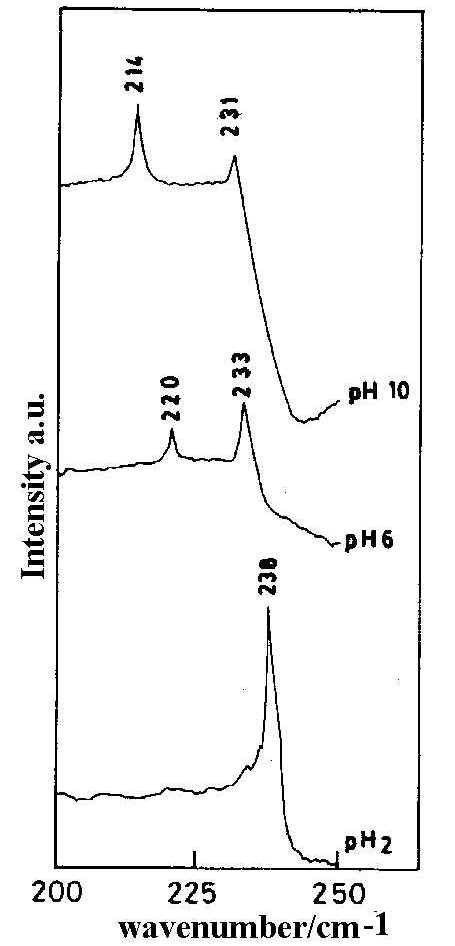
Figure 7. SERS spectra of hypoxanthine (low frequency range) at different pH. [laser power 0.25 W, l = 514.5 nm]
From Figure 6, it is seen that as the pH of the solution is increased from 2 to 6 there is no sharp change in the peak position of the 724 cm-1 band but its relative intensity drops substantially. Another band centred at 1337 cm-1 assigned to ring skeletal vibrations shows some interesting changes. At pH 2 this band is strongly enhanced but with an increase of pH the peak intensity becomes relatively weak. At pH 8 a drastic shift of the ring breathing mode from 724 cm-1 to 746 cm-1 is observed accompanied by a relatively weak ring skeletal vibration. This sudden change in the SER spectra may be attributed to the change in the orientation of the molecule on the silver substrate due to pH variation. At still higher pH (pH 10) the overall spectral quality detoriates and the very weak ring skeletal vibration is shifted to 1320 cm-1.Interestingly, at this pH the relative intensity of the ring breathing mode also drops substantially.
Under acidic conditions (pH 2) the enhancement of the ring skeletal (1337 cm-1 ) and ring breathing (724 cm -1) vibrations suggests that the HX molecule is adsorbed on the metal surface through the N3 atom with the molecular plane lying perpendicular to the silver surface. As the pH is increased, the Raman intensity of the skeletal mode as well as ring breathing one drops substantially indicating a configurational change of the molecule. Hence at alkaline pH where HX exist in the enol form, the molecule is oriented nearly parallel to the silver surface via nonbonding electrons of N1 and O6 atoms and also possibly through the N3 and N7 atoms. Infact, the blue shift and the decrease in enhancement of the ring breathing mode also suggest the nearly parallel orientation of the molecule.
Thus the change in pH of the sol results in tautomeric and configurational change of the HX molecule.
pH dependent SER spectra of HQ
We have also studied the effect of pH variation on the SER spectrum of 8-hydroxy quinoline [HQ] adsorbed on to a silver hydrosol. Our earlier discussion on the effect of pH variation on the SERS intensity of 4-PCA and HX suggests that the SER spectra at varied pH can efficiently identify the form of the molecule at the interfacial environment. However, in the present case we show that with pH variation, two types of surfaces, namely chlorinated and bare silver surfaces, predominates for the adsorption process and this is reflected in the SERS spectra of the HQ molecule.
The chemical structure of 8-hydroxyquinoline (HQ) is shown in Figure 8. HQ belongs to the Cs point group and consequently 33 planar (A/ species) and 15 non-planar (A//species) fundamental vibrations are expected to appear both in the Raman and in the FTIR spectra. Compared to the solid state Raman spectra which shows 28 bands, the solution spectra (0.1 M) is rather poor and only 10 bands are observed. These Raman and the IR vibrational bands are convincingly assigned and have been reported in the literature [33,34].
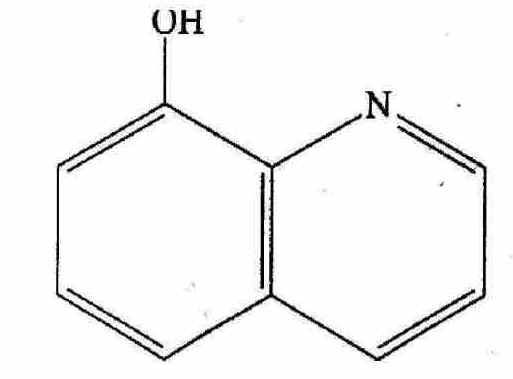
Figure 8. Schematic representation of 8-hydroxy quinoline molecule.
The SER spectra of HQ adsorbed on a silver hydrosol, as shown in Figures 9 and 10, contain twenty bands whose intensity and position changes with the pH of the solution. As the pH value increases from 2 to 10.5, some pairs of Raman bands show intensity reversal. For example, the doublet at 495 and 514 cm-1 representing out-of-plane ring deformations and the ring stretching modes at 1566 and 1593 cm-1, the 495 and 1566 cm-1 bands show enhanced intensity compared to the 514 and 1593 cm-1 bands as the pH value increases. Similarly, of the features at 715 and 726 cm-1 representing ring breathing modes and of the 1363 and 1400 cm-1 bands assigned to ring stretching modes respectively, the 726 and 1363 cm-1 peaks are more intense at higher pH compared to the bands at 715 and 1400 cm-1. With change of pH, as no new band appears to suggest structural changes of the HQ molecule, we may attribute the spectral variations to orientational changes of the molecule on the silver surface. Alternatively, such intensity change could be attributed to the change in the adsorption sites with pH [35]. Now, as we have mentioned earlier, the pH variation is performed by adding dilute HCl or NaOH solutions. At pH 2 and 4 the Cl– ion concentrations are 1 x 10-2 M and 1 x 10-4 M respectively. At nearly neutral pH (pH ~7) all the Cl– ion in the medium is eventually neutralised by the Na+ ions from NaOH. For higher pH values, it is the OH– concentration rather than H+ ions which determine the pH variation. There may be both bare and chlorinated sites on the colloidal silver surface and as the relative concentration of these two types of sites changes with pH, the intensities of the bands representing vibrations of molecules occupying two different sites also changes. At lower pH , there will be more chlorinated sites whereas at higher, there will be more bare silver sites. The Raman bands of molecules located at the chlorinated sites will decrease in intensity whereas the Raman bands of molecules sited on at bare silver atoms will increase in intensity as the pH value increases. As we have discussed in a following section on the estimated enhancement factors of the SERS bands, adsorption of the molecules at bare and chlorinated silver surface is likely to be responsible for pH dependence of SERS spectra. The vibrational modes of the HQ molecule adsorbed at the chlorinated and bare silver sites are similar but they may differ in frequency and in intensity. The intense ring modes show up the difference between the molecules adsorbed at the two different sites clearly. Weak broad bands show little difference. Orientation of the molecules at the two sites may be such that some specific modes also remain unchanged.
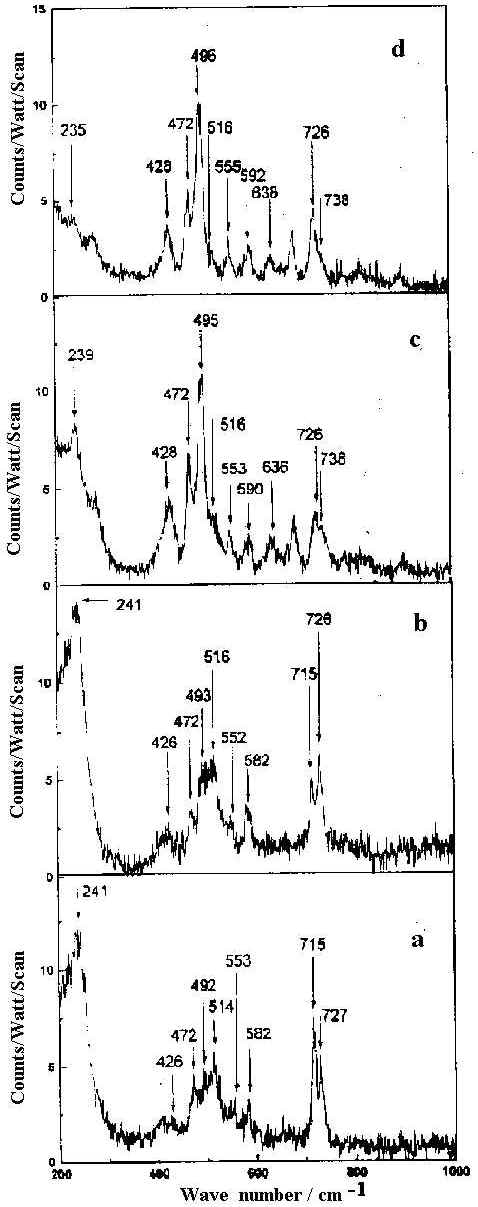
Figure 9. SERS spectra of 8-hydroxy quinoline (1 x 10-4 M) in 200-1000 cm-1 range at pH
a) 2 b) 4 c) 7 d) 10.5.
All spectra are calibrated in counts/watt/scan.
[laser power 0.1 W, l = 514.5 nm]
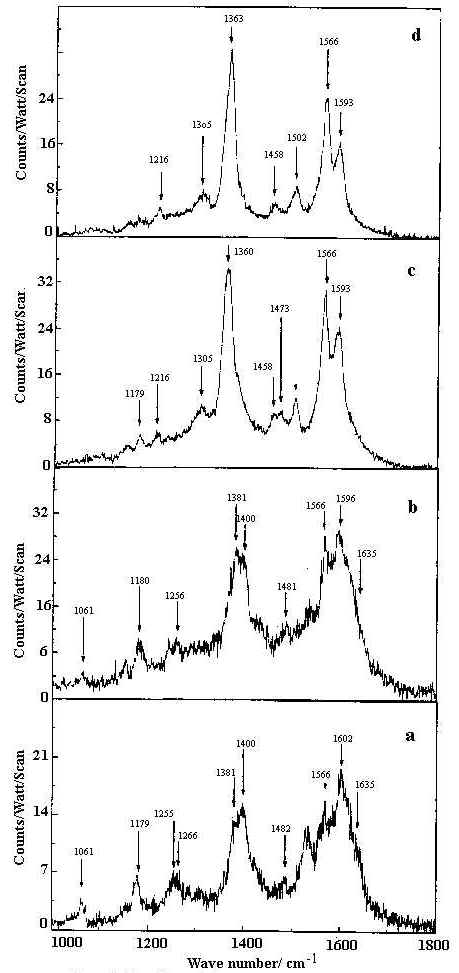
Figure 10. SERS spectra of 8-hydroxy quinoline (1 x 10-4 M) in 1000-1800 cm-1 range at pH
a) 2 b) 4 c) 7 d) 10.5.
All spectra are calibrated in counts/watt/scan.
[laser power 0.1 W, l = 514.5 nm]
The band at 241 cm-1 is intense but broad at pH 2. At pH 4 sharpening of this band occurs. At pH 7 the intensity of this band drops and is shifted to 239 cm-1 . At still higher pH (pH 10.5) this band appears as a weak broad hump at 235 cm-1. It is reported that both Ag-Cl and Ag-N vibrations occur at very close frequencies in this region [7]. Thus at pH 2, where the origin of the SER spectrum is principally due to the the chemisorption of HQ on the bare and Cl– ion adsorbed silver surfaces, it is quite plausible to think that both Ag-Cl and Ag-N vibrational contributions give rise to the intense band at 241 cm-1. At pH 4, as the Cl– ion concentration is reduced in the medium, the greater contribution is expected from the Ag-N stretching mode at 241 cm-1 compared with the Ag-Cl vibrations. At pH 7 and 10.5 where the origin of SER spectra is mainly due to the chemisorption of HQ molecules on chlorine free silver surface, the 239 cm-1 mode at pH 7 and 235 cm-1 mode at pH 10.5 correspond solely to the Ag-N stretching vibration of silver-HQ system. Thus the appearance of Ag-N stretching vibrations in the whole range of pH indicate that the molecule is adsorbed on the silver surface through the nonbonding electrons of the nitrogen atom of HQ molecule.
The other striking feature of the SER spectrum is the variation in Raman intensity of bands near 428 and 472 cm-1 with the increase of solution pH. Both these bands are very weak at pH 2 and 4 but increase in intensity towards neutral and alkaline pH. Of these the 472 cm-1 band is assigned to the in-plane C-O bending vibration, while 428 cm-1 band corresponds to the out-of-plane ring deformation.
Orientation of the adsorbed molecule on the metal surface can be estimated from the enhancement of the relevant Raman bands following the surface selection rule based on image dipole theory as predicted by Creighton et al. [9] and Van Duyne [36].
From the SER spectra it is clearly seen that the in-plane C-O bend at 472 cm-1, out-of- plane ring deformation at 496 cm-1, ring breathing at 726 cm-1 and ring stretching modes at 1363 and 1593 cm-1 are significantly enhanced. Such enhancement, however does not show any significant dependence on the sol pH. This indicates that the HQ molecule does not undergo any change in orientation with the variation of the solution pH. Also the appearance and enhancement of the in-plane and the out-of-plane modes in the SER spectra indicate that the molecule is oriented neither flat nor vertical to the metal surface but is tilted. The appearance of the Ag-N stretch confirms that the molecule is co-ordinated to the silver surface through the non bonding electrons of the nitrogen atom. In fact, the frequency shift and change in relative intensity of the Raman bands at neutral and alkaline pH may be attributed to the chemisorption of the HQ molecule on the bare or chlorinated silver surface.
Thus we show that the pH dependent SER study can be efficiently and successfully utilised to identify the protonated, deprotonated and hydrated state of the 4-PCA molecule. It can also indicate tautomeric and orientational changes in the HX molecule and enable us to understand the effect of the bare and chlorinated silver surfaces on the SERS spectra of HQ molecule.
Acknowledgement
The authors express their thanks to DST, Government of India for financial support.
References
- R. P. Van Duyne, in Chemical and Biological Applications of Lasers, C.B. Moore (Ed.), Vol. 4, Chap. 1., Academic Press, New York, 1979.
- R. K. Chang and T. E. Furtak (Eds.), in Surface Enhanced Raman Scattering, Plenum Press, New York, 1982.
- R. P. Cooney, M. R. Mahoney and A.J.McQuillan, in Advances of Infrared and Raman Spectroscopy, R. J. H. Clark and R. E. Hester (Eds.), Vol.9, p.188. Heyden, London, 1982.
- J. Gersten and A. Nitzan, J. Chem. Phys. 73, 3023 (1980).
- T. E. Furtak and S. H. Macomer, Chem.Phys. Lett., 95, 328 (1983).
- J. R. Lombardi, R. L. Brike, T. Lu and J. Xu, J.Chem.Phys., 84, 4174 (1986).
- M. Muniz-Miranda, N. Neto and G. Sbrana, J.Phys. Chem., 92, 954 (1988).
- J. F. Arenas, I. Lopez Tacon, M. S. Wooley, J. Cotero and J. I. Marcos, J. Raman Spectrosc., 29, 673 (1998).
- J. A. Creighton, C. G. Blatchford and M. G. Albrecht, J. Chem. Soc., Faraday Trans., 75, 790 (1979).
- P. C. Lee and D. J. Meisel, J. Phys. Chem., 86, 3391 (1982).
- S. Sanchez-Cortes and J.V. Gracia-Ramos, Vib. Spectrosc., 4, 185 (1993).
- K. U. Von Raben, R. K. Chang and B. L. Laube, Chem.Phys. Lett., 79, 465 (1981).
- W.S Sutherland and J.D. Winefordner, J. Colloid Interface Sci., 99, 270 (1984).
- L. E. Camafeita, S. Sanchez-Cortes and J. V. Gracia-Ramos J. Raman Spectrosc., 26, 149 (1995).
- J. Neddersen, G. Chumanov and T. M. Cotton, Appl. Spectrosc., 47, 1959 (1993).
- M. Prochazka, P. Mojzes, J. Stepanek, B. Vlckova and P. Y. Turpin, Anal. Chem.,69, 5103 (1997).
- E.Vogel, W. Kiefer, V. Deckert and D. Zeisel, J. Raman Spectrosc., 29, 693 (1998).
- K. Abe and M. Hirota, Bull. Chem. Soc. Jpn., 50, 2028 (1977).
- M. Paris, G. Thomas and J. C. Merlin, Bull. Soc. Chem. Fr., 707 (1961).
- W. Kemp, Organic Spectroscopy , ELBS, Hong Kong, 1986.
- K. A. Bunding and M. I. Bell, Surf. Sci., 118 ,329 (1983).
- K. Mukherjee and T. N. Misra, J.Chem. Soc., Perkin Trans. 2., 2227 (1996).
- T. Nonaka, T. Kato, T. Fuchigami and T. Sekine, Electrochim. Acta.., 26, 887 (1981).
- W. S. Oh, M. S. Kim and S. W. Suh, J.Raman Spectrosc., 18, 253 (1987).
- S. Sanchez-Cortes and J. V. Garcia-Ramos, J. Raman Spectrosc., 21, 679 (1990).
- M. Mathlouthi and A. M. Seuvre, Carbohyd. Res., 146, 1 (1986).
- G. Varsayani, Vibrational Spectra of Benzene Derivatives, Academic Press, New York, 1969.
- K. Mukherjee and T. N. Misra, J. Raman Spectrosc., 27, 595 (1996).
- D. B. Boyd and W. L. Lipscomb, J. Theor. Biol., 25, 403 (1969).
- J.Stewart, MOPAC Program 455, Quantum Chemistry Program Exchange, University of Indiana, Bloomington, IN. ,
- M. J. S. Dewar, E. G. Zoebish, E. F. Healy and J. J. Stewart, J. Am. Chem. Soc.,107 3902 (1985).
- M. J. S. Dewar and W. Thiel, J. Am. Chem .Soc., 99, 4899 (1977).
- S. L. Srivastava, M. Prasad and Rohitashava, Spectrochim.Acta., 40A, 681 (1984).
- B. Marchon, L. Bokobza, and G. Cote, Spectrochim.Acta., 42A, 537 (1986).
- M. Takahashi, H. Idetsuki and M. Ito, Surf. Sci., 232, 346 (1990).
- C. S. Allen and R. P. Van Duyne, Chem.Phys. Lett., 63, 455 (1979).
REF: J.Chowdhury, M. Ghosh, K.M. Mukherjee and T. N. Misra Int. J. Vib. Spect., [www.irdg.org/ijvs] 4, 2, 7 (2000)