Highly-sensitive ATR Raman spectroscopy using Surface-Plasmon-Polariton
Masayuki Futamata
Joint Research Center for Atom Technology (JRCAT)
National Institute for Advanced Interdisciplinary Research (NAIR)
1-1-4 Higashi,
Tsukuba 305-8562, Japan
E-mail: futamat [at] jrcat.or.jp
Introduction
Although the electrode/electrolyte interface is of fundamental importance in energy conversion, material synthesis, sensing, corrosion inhibition and catalysis, the reactions involved have primarily been characterised using electrochemical techniques such as cyclic voltammetry. These methods although of very high sensitivity can only provide relationships between current and potential or surface charge for the whole electrode. As a consequence, our understanding of these solid/liquid interfaces is not nearly so highly developed as that in solid/gas systems where a wide range of molecular level tools are available.
Recently, progress has been made thus electrochemical systems were studied at the atomic level using scanning probe microscopy [1], surface X-ray scattering [2] and in the use of the Electrochemical Quartz Crystal Microbalance. However, valuable though they may be, they fail to provide information on the nature of adsorbates at electrodes or of their interaction with the electrode surface. Vibrational spectroscopy can provide this information particularly because highly sensitive methods have become available.
In this paper, a new technique of highly-sensitive ATR (attenuated total reflection) Raman spectroscopy using Surface Plasmon Polariton (SPP) is described. In such that there is a prospect that in the near future dynamic processes involving adsorbates at working electrode surfaces will be elucidated at an atomic level.
As has been pointed out elsewhere in this edition of the Journal, Raman spectroscopy is a scattering process, obeying a different selection rule from infrared spectroscopy and provides complimentary information on molecular vibrations. Additionally, the symmetry of molecules sorbed to surfaces affects the relative intensity of bands in the spectra and the latter can be used to define the orientation and interaction of these molecules with the electrochemical substrate as a function of potential.
Unfortunately, Raman scattering is a two photon process of low probability [scattering cross sections are typically = 10-30cm2/molecule] i.e., it is of far lower intensity than single photon processes such as IR absorption where is closer to10-20cm2/molecule or electron energy loss spectroscopy where is even larger at 10-16cm2/molecule. Accordingly Raman spectroscopy has insufficient sensitivity to characterise dynamic processes of monolayer adsorbates. Therefore, we have studied to improve the sensitivity by combining a new scattering process and a highly efficient spectrometer.
In this article, the characteristics of ATR Raman spectroscopy using SPP over smooth metal surfaces are reviewed and compared with SERS on roughened electrodes. The experimental arrangements are described and the results discussed against a theoretical background. Finally, the ATR Raman spectroscopy combined with scanning near field optical microscope is reported and its potential in the future is discussed.
ATR-SPP Raman spectroscopy using PSPP on smooth metal surfaces
SERS using localized SPP (LSPP) on roughened metal surfaces is known to give an enormous enhancement of 104-106, which is larger than the enhancement of 102-103 by propagating SPP (PSPP) on smooth metal surfaces as described in this section. However, there are substantial difficulties to utilize SERS on the rough surface as an analytical tool.
Two distinct mechanisms have been proposed to explain the observed result [3] one due to electromagnetic enhancement (EM [4]) and the other chemical enhancement (CE). In the EM mechanism, the localized SPP (LSPP) on roughened or colloidal metal surfaces enhances the electric field |E|at the surface [4], which decays over a long-range according to the relationship of |E|2 µ (a/r)10 (a: radius of a hemisphere, r: distance of the adsorbates from the Ag surface) for a hemispherical Ag particle [5]. Raman scattering is enhanced effectively only by the excitation of the LSPP longitudinal for prolate metal particles [6]. The other evidence for the EM mechanism is that the excitation profile maximum progressively moves to longer wavelength with increasing the aggregation. However, detail relationships between the shape and size of each colloidal particle and the enhancement in SERS is not yet at hand. For instance, single colloidal particle in resonance with the excitation wavelength has a particular size distribution much narrower than the theoretical prediction, suggesting a size sensitive mechanism [7]. Then, we have studied SERS on Ag colloidal particles using various preparation methods, AFM imaging, absorption and Raman spectroscopy. In addition to the enhancement of 105 – 106 for various adsorbates including DNA bases, we find higher enhancements for lower coverage of adsorbates at each Ag particle, e.g. 105 for 10-7 M and 108 for 10-12 M of dye molecules incubated. Moreover, time-dependent changes in the Raman band intensity (blinking) is observed, suggesting that there exists SERS active sites with vast enhancements and that migration of the adsorbates occurs on the Ag colloidal particle [34].
The CE mechanism [3] was proposed based on the following three observations. (a) Metal films evaporated at low temperatures or on CaF2 underlayers contain atomic-scale roughness (also named in some sources as SERS active sites) which yields much larger enhancement but only over short-ranges (only for the first layer) characteristic in contrast to the long-range effect in the EM mechanism. The active site is passivated by dosing atomic oxygen onto the surface. (b) The enhancement depends on molecules and their vibrational modes, because the mechanism is based on the intrinsic charge transfer interaction between metal and molecular orbital of adsorbates. (c) The peak potential, at which the Raman band intensity shows the maximum in electrochemical environments, shifts with changing the excitation wavelength. The difficulty in discussing this mechanism is that the active site at the atomically roughened surface has not been elucidated at the atomic level. It was also been reported that even on Ag(111) surfaces pyridine has a significant interaction to give a charge transfer band as observed in EELS [8]. These issues for both mechanisms must be solved if one is to confirm single molecule detection with SERS [7, 9]. An alternative method is to use an ultra thin metal film evaporated onto a particular substrate at room temperature, providing the SERS active surfaces promising in the characterization of the adsorption. These surfaces consist of particularly well-arranged particles with certain surface orientation (facet), thus they are similar to single crystal surfaces to some extent. For instance, the Ag and Au thin film evaporated on to Si or ZnSe prisms consists of ellipsoidal particles of several tens of nanometer in diameter, and with (111) orientation [10]. This surface can be confirmed by electrochemical measurements. The cyclic voltammograms observed are essentially similar to the single crystal surface of Au(111) for various electrochemical processes, e.g. adsorption of sulfate ions or the underpotential deposition process of Cu [10].) In the future, when Scanning Near-field Optical Microscopy (SNOM) is combined with Raman spectroscopy, which will be described in the later section, we will have molecular level spatial resolution and the mechanism of SERS will be elucidated.
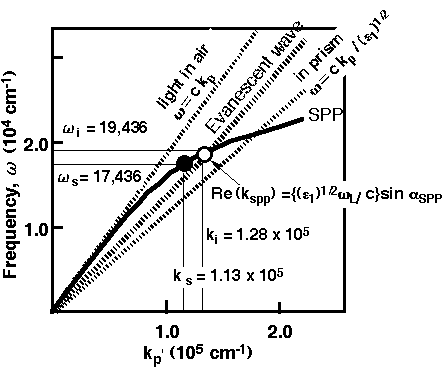
Figure 1. Dispersion relation of the SPP. Dotted lines denote the relation of the light in air (left) or in prism (right, see the text).
In- and out-coupling of the SPP with light occur at the crossing points
(O,• ) [13]
At present, it is particularly hard to evaluate how the large enhancements observed for certain rough surfaces originates from these two mechanisms, and thus hard to discuss quantitatively the adsorbed state. Therefore, it is valuable to use the PSPP, since such surface roughness is not necessary, and thereby one can apply this to the bulk single crystal surface allowing us to discuss the observed data combined with other excellent techniques such as SPM or SXS. The bulk plasmon frequency for a collectively excited state of electrons in a metal is in the UV region and can be excited by UV light. Surface plasmons interact with p-polarized light unlike to the bulk plasmon due to the boundary condition at the surface [11, 12]. This was exploited to excite the PSPP with light by Otto [11] and independently by Kretschmann [12] in the early 70’s. As shown in Figure 1 [13], the wave vector for the PSPP parallel to the surface (kp) lies to the right from the light in air (kp>w/c). Thus, the PSPP cannot be excited with light by the direct irradiation of the metal surface. However, the wave vector of the light is increased using the ATR method or by using a grating. For instance, the evanescent field with its wave vector of the light in the prism is permeated into air, when the light is incident upon the spherical prism base with an angle (ai ) above the critical angle for total reflection: kp = (w/c)e1½ sin ai, e1: the dielectric constants of the prism. When the incident angle is adjusted (ai = aSPP), the wave vector kp accords with that of the PSPP, i.e., the energy and wave vector of the light accord with those for the PSPP. Accordingly, one can excite the PSPP with light at the crossing point of the dispersion curves in Figure 1. Experimentally, this is clearly shown by the sharp minimum in reflectivity at the resonance angle. Figures 2a and 2b show the optical setup needed to excite the propagating surface plasmon polariton. In addition to the angle of incidence, the gap size between the metal surface and prism base can be optimized in the Otto configuration for the most efficient utilization of the SPP [12] (Figure 2a). Similarly, the thickness of the metal film directly evaporated onto the ATR prism base is optimized in the Kretschmann configuration (Figure 2b). Adsorbates on the metal surface will affect the dispersion relation of the PSPP, hence the dielectric constants and/or the thickness of the adsorbates can be evaluated through slight changes in the resonance angle and/or in the width of the reflectivity dip. Recently, chemical sensors, biosensors [14] and even a surface plasmon microscope [15] using this phenomenon have been developed and applied in a variety of fields.
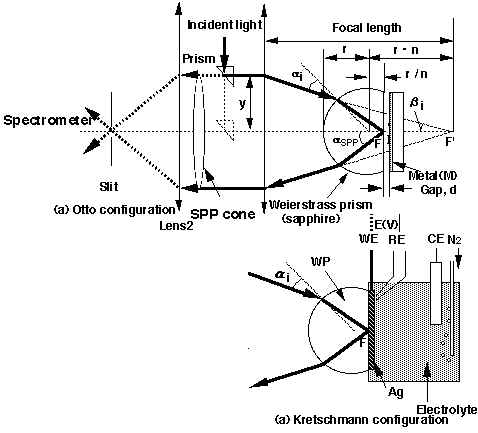
Figure 2. ATR-Raman configuration to utilize in- and out-coupling of the SPP on the metal (M): (a) in Otto[21-24], and (b) Kretschmann configuration [25]. Prism position (y) is moved perpendicular to the optical axis to change the incident angle ai( = arcsin (n sin bi)) to the Weierstrass prism (WP, r: radius, n: refractive index). The scattered light within the SPP cone goes parallel to the optical axis beyond the first objective (L1, f1: focal length), and is focused on the entrance slit (ES) of the monochromator with the second objective (L2). Here wL: incident light, d: gap size between the prism base and metal surface, WE: working, CE: counter and RE: reference electrode for potential control.
The enhancement of the Raman signal from adsorbates using the SPP has also been discussed with respect to the mechanism of SERS. Ushioda et. al. have reported that the observed Raman intensity can be fitted well by assuming that the enhanced scattering process is mediated by the SPP, and that the enhancement factor for the C-H stretching band in MeOH on a Ag surface is 4 x 104[16]. They have also reported theoretical formulas for calculation [17-19] of the enhancement and the decay length (ca. 150 nm) of the SPP field using a MgF2 spacer over a silver film [17]. The SPP field strength decays from the silver film surface to the air as in the evanescent field. (The decay length means the distance where the SPP field strength decays down to 1/e of the silver surface.) Pettinger et al. applied this method to the electrode surface [20].
All of these are based on the Kretschmann configuration, and the following are insufficient from an analytical point of view: (1) utilization of out-coupling (Figure 1), which is a reciprocal process to in-coupling [35], and leads to an additional enhancement, (2) how the resonance condition is affected by both the dielectric properties and the thickness of the adsorbates, and (3) relevance to different metals. Moreover, the Otto configuration possesses several advantages e.g., it is feasible in application to the bulk single crystal surface and additional enhancement arises from interference of the scattered light in the gap. Thus, in order to establish ATR-SPP Raman spectroscopy as an analytical tool, we have studied details of the SPP resonance conditions and determined the Raman band intensity as a function of the dielectric properties and thickness of the constituents including the metal, adsorbates, media, and the prism itself at various wavelengths, primarily using the Otto configuration [13, 21-26].
Experimental set up
Figure 2 shows the optical setup to excite the SPP with the Otto (a) and Kretschmann configurations (b). The light is incident and collected at the same side of the prism to utilize both the in- and out-coupling. The angle of incidence to the prism base is tuned by moving a small prism (p) perpendicular to the optical axis (y). When the angle is in accord with the resonance angle (ai = aSPP), the SPP is excited at the metal surface. Because the excited SPP is elastically scattered by microscopic defects, steps or roughness, where it has a wave vector for every direction with respect to the surface [21]. Strong Raman signals from adsorbates are emitted towards the prism side (incident side) at the resonance angle for the out-coupling, which is slightly different from the incident beam, because the energy and wave vector of the SPP shifts due to the Raman scattering process of the adsorbates. Prominent Raman scattering is emitted into the air side of the prism only for rough surfaces [13, 21-26]. Thus for a smooth Ag surface the predominant Raman signal is scattered within a cone originating at the prism base with the resonance polar angle and an arbitrary azimuth angle. In order to collect all the scattered light within the core, we use an objective with a large acceptance angle (a small F value). Furthermore, the resonance angle for the electrode/electrolyte interface is much larger than in air, for instance the acceptance angle of F/0.7 is ca. 32-33° , which is smaller than the resonance angle of 35° at 647.1 nm for ZnSe/H2O interface. Therefore, we reduce the apparent scattered angle with the Weierstrass prism [27],which is longer than a half-spherical prism by r/n where r = radii and n: refractive index, see also Figure 2). A two-dimensional suspension in air [21] or Teflon bellows in solution was used to make the metal surface parallel to the prism base. This enables us to optimize the gap size for the irradiated area of the sample. Figure 3 shows the direct image of the scattered light taken on printing paper placed just backward of the objective 1. The SPP cone is clearly observed as the outside ring, while the interference fringe is seen in the inside region raised from the out-coupling by the slight roughness of the metal surface. The centre of the cone repeatedly becomes bright and dark with every step of the gap size Dd = lL/4 according to the interference condition of 2dn cosq = mlL (m = 0, 1, 2, .., n: refractive index of media, d: gap size, q : scattering angle, lL: wavelength of the incident light). The gap sizes observed with this method agree well with the theoretical values as described below [13, 21-24]. The Otto configuration has not been used as much as the Kretschmann configuration due to the experimental constraints in controlling the gap size. Thus, the device developed here is essential to utilize the Otto configuration, which is relevant to bulk single crystal surface and to discuss the orientation of adsorbates. For the electrochemical interface, the diffusion limitation in a conventional thin layer solution cell as used for example in Infrared Reflection Absorption Spectroscopy (IRAS) [28] or SERS can be a problem. However, it is overcome using the Kretschmann configuration, since the thickness of solution layer has no limitations in the cell containing a counter electrode, and reference electrode (see Figure 2b). Thus, ATR Raman spectroscopy is efficiently utilized by employing an optimum electrochemical configuration for a particular sample system. Our sample system contains the Ag, Au, Cu, Ni or Pt films evaporated onto BK-7 glass substrates, ZnSe or the sapphire prism base, and copper phthalocyanine (CuPc), pyridine (Py) or self-assembled monolayers (SAM) as adsorbates.
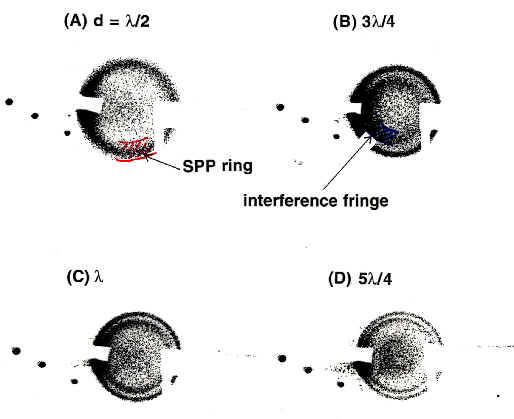
Figure 3. The SPP cone and interference fringes at
various gap sizes d [13,21]
Theoretical evaluation
The local field intensity at the metal surface in the prism/air gap/metal system can be evaluated using the Fresnel equation derived from Maxwell’s equations and the boundary conditions [24]. The local field intensity at SPP resonance is normalized to that under the external geometry. As shown in Figure 4, the largest enhancements occur for the larger real values and small imaginary values of the dielectric constants for the metal. For instance, since Ag has the large absolute value > 10 for the real part (Ree) and a quite small value for the imaginary part (Ime) of < 1 in the visible wavelength region, an enhancement of ca. 300 is predicted (Figure 4a). Note that the enhancement factor does not necessarily decrease with increasing imaginary part component for |Ree|> ca.10 see Figure 4b. Thus, for example, Pt or Ni will give a significant enhancement of ca. 30. In addition, the enhancement increases monotonically with increasing imaginary component for a specimen with a small real one e.g., -1.0. Since enhancement of 30 reduces the measuring time by a factor of 900, the experiment is still promising for the transition metals. Moreover, these values result from the in-coupling of the SPP. A further enhancement is obtained from out-coupling, e.g., the induced Raman dipole perpendicular to the surface provides an additional enhancement of ca. 50 due to this out-coupling. This evaluation supports the relevance of ATR-SPP Raman spectroscopy to transition metals most of which have higher intrinsic damping than in Ag. Therefore, we tried to apply the principle to the Pt and Ni surfaces. The formulae for the scattering intensity under SPP resonance conditions contain the dielectric constants, and the Raman tensor elements of the adsorbates. Thus, we can evaluate the orientation of the adsorbates from comparison between the observed and theoretical enhancement factors and the anisotropy of the SPP field.
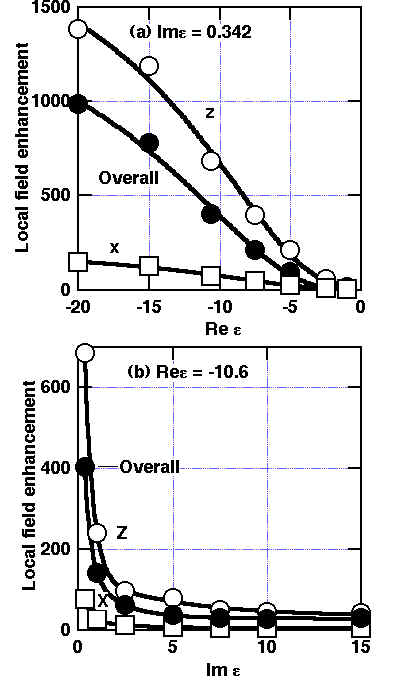
Figure 4. Localised electric field enhancement G due to the SPP excitation [24]:
(a) with fixed imaginary part (Im emetal = Im eAg
(at 514.5nm)=0.342),
(b) with fixed real part (Re emetal = Re eAg (at 514.5nm) = -10.60)
Results for various metals
(Au, Ag, Cu, Ni and Pt)
At first, the ATR reflectivity for the sapphire prism/air gap/CuPc/ Ag system was measured using a 514.5 nm wavelength. Sharp and deep reflectivity dips were observed at ca. 36.8 ° (in air) only for p-polarized light, indicating that excellent energy transfer of > 95 % from the light into the SPP (see Figure 5) should occur [13, 21]. The reflectivity shows a minimum at a gap size of ca. 3/4lL (Figure 6). The resonance angle and the optimum gap size significantly shift with increasing CuPc thickness due to the dielectric properties of CuPc as shown in Table 1. Furthermore, the Raman band intensity is maximised itself at the reflectivity minimum (Figures 5, 6). Since the observed resonance conditions are in good agreement with theoretical predictions based on the Fresnel equations for the prism/gap/CuPc/Ag system, the SPP was excited with the method described here and we obtained the enhancement factors in Raman band intensity. As shown in Figure 7, for Au as well as Cu, quite broad reflectivity dips were observed at shorter wavelength of excitation than in Ag due to the larger imaginary component of the dielectric constants. However, at longer wavelength beyond ca. 600 nm, the imaginary part intrinsically decreases and the reflectivity shows sharp and deep minima.
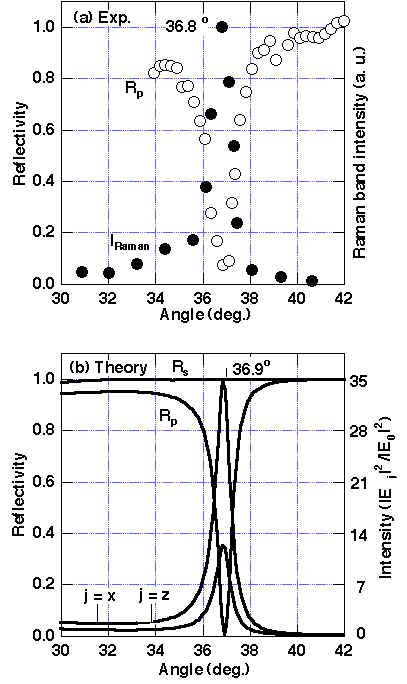
Figure 5. Reflectivity R at p-polarization and unpolarized Raman band intensity I from CuPc at 1530cm-1 in the Weierstrass prism/air gap/CuPc (5.8nm)/Ag system for various incident angles [13]:
(a) observed (R:O, I:•) and
(b) theoretical curves, where |Ej|2/|E0|2 is the squared electric field parallel (j=x) or perpendicular (z) to the surface normalized to that of the incident field (j=o). The gap size dopt is 3.5lL/4 (exp.) or 3.5lL/4 (theory).
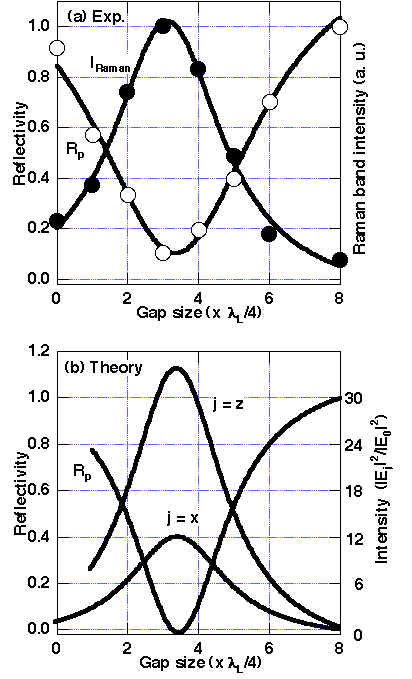
Figure 6. Reflectivity R at p-polarization and unpolarized Raman band intensity I from CuPc at 1530 cm-1 in the Weierstrass prism/air gap/CuPc (5.8nm)/Ag system for various gap sizes [13].
(a) observed (R:O, I:•) and (b) theoretical curves. The incident angle is 36.8 ° (obs) or 36.9 ° (theory).
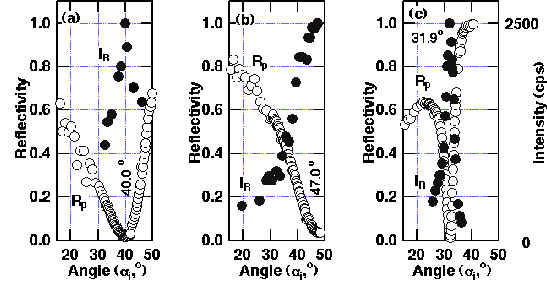
Figure 7. Reflectivity R (O ) at p-polarization and unpolarized Raman band intensity I ( •) from CuPc at 1530 cm-1 observed in the Weierstrass prism/air gap/CuPc(5 nm)/Au systems for various incident angles and excitation wavelength ((a) 457.9 nm, (b) 514.5 nm and (c) 632.8 nm) [23]. The gap size is < lL/4 (at 457.9 nm, and 514.5 nm) or 3lL/4 (at 632.8 nm).
The Raman band intensity changes as a function of thickness of CuPc, and showed a maximum enhancement of 400 at 2.0 – 2.5 nm for CuPc (see Table 1 [13]). If the adsorbate has an intrinsic damping, the enhancement should monotonically decrease with increasing thickness. Note that the enhancement for thinner CuPc layers is rather modest. This is due to an additional enhancement originating in the external geometry, and results from an increased out-coupling caused by the roughness in the thinner CuPc films. This suggestion is supported by the fact that the observed intensity and the theoretically derived values are in good agreement under the propagating SPP resonance conditions. The decay of the SPP field is substantially dependent on internal damping in the CuPc layer. We find that the decay length is only ca. 16 nm [13], significantly smaller than the value of ca. 150 nm estimated by Ushioda et al for a MgF2 layer [17]. Since most adsorbates of interest have an absorption in the visible, the decay length to guaranteed to be small enough that the outside bulk species supplies a negligible contribution to the observed Raman spectra in electrochemical environments. As the dielectric dispersion of Ag is quite small, both slight changes in the resonance conditions and also in the enhancement factors for different excitation wavelengths result from those for CuPc. Although the SPP can be efficiently excited for both Au and Cu at longer wavelengths, the enhancement is not large because overlap with the p-p * absorption increases the intrinsic damping of the SPP field in the CuPc layer (Table 2). In fact, much larger enhancement of ca. 100 are obtained for p-nitrothiophenol (PNTP) – self-assembled as monolayer (SAM) films over Au. We used metal films of Ag, Au and Cu evaporated under high vacuum and consisting of ellipsoidal particles with a diameter of several tens of nm. The CuPc molecules are randomly oriented to the metal surfaces in these systems. As it turns out the enhancement factor calculated for random orientation agrees well with the observed values as shown in Table 2. An additional enhancement by a factor of 4 was obtained in water media. This arises from increased out-coupling in media with larger dielectric constants as predicted by theoretical calculations [13].
| Thickness (nm) |
aSPP (° ) | a dopt b (´ lL/4) | FWHM (°) c | Enhancement d | |||
| obs. | calc. | obs. | calc. | obs. | calc. | ||
| 0 | 36.3 | 36.2 | 4.0 | 4.5 | 0.80 | 0.46 | |
| 1.5 | 36.4 | 36.5 | 4.0 | 4.5 | 1.28 | 0.46 | 146 |
| 2.0 | 36.5 | 36.5 | 4.0 | 4.0 | 0.82 | 0.48 | 400 |
| 2.5 | 36.6 | 36.6 | 4.0 | 4.0 | 1.05 | 0.50 | 399 |
| 4.0 | 36.7 | 36.7 | 3.5 | 3.5 | 1.01 | 0.72 | 303 |
| 5.8 | 36.8 | 36.9 | 3.5 | 3.5 | 1.22 | 0.80 | 275 |
| 11.7 | 37.6 | 37.6 | 3.0 | 2.5 | 1.77 | 1.72 | 149 |
| 15.0 | 38.4 | 38.2 | 3.0 | 2.5 | 2.18 | 1.90 | 121 |
Table 1. Resonance conditions to excite the SPP in prism/air gap/CuPc/Ag at 514.5 nm for different thickness of adsorbates [13].
a Resonance angle for in-coupling
b Optimum gap size, where lL is the wavelength of the excitation light.
c Full width at the maximum of ATR spectra.
d Raman band intensity of 1530 cm-1 from CuPc under the SPP resonance normalized to that for external reflection geometry.
| Wavenumber (cm-1) |
Enhancement factor* | |||||||
| Au† | Cu† | Ag | Ag H2O** |
Pt 3.5# |
Pt 8.6# |
Ni 4.5# |
||
| 1535 | A1g | 49.9 | 22.6 | 275 | 1228 | 288 | 164 | 24.5 |
| Theory*** | ||||||||
| Random | 25 | 24 | 354 | 1080 | 40 | 30 | 26.5 | |
| Parallel | 44 | 37 | 5710 | 9340 | 250 | 243 | 270 | |
| Perpendicular | 13 | 14 | 29 | 53 | 7.5 | 4.8 | 4.5 | |
| 1457 | B1g | 41.6 | 20.5 | 288 | 1305 | 291 | 163 | 23.7 |
| 1346 | A1g | 55.4 | 22.5 | 286 | 1322 | 261 | 180 | 21.7 |
Table 2. Enhancement factor for Raman band intensity of CuPc
(t = 6 nm) on various metals under the SPP resonance [13, 22-24].
* Normalised to the intensity for the external scattering geometry at the same angle of incidence.
** In H2O solution in other case in air.
*** Theoretical values for random, parallel or perpendicular orientation of CuPc to the Pt surface.
† At 632.8nm in other case at 514.5nm.
# Thickness of CuPc in nm.
ATR-SPP Raman spectroscopy was applied to the transition metals with the configuration prism/air gap/CuPc (t = 8.6 nm)/ Pt as shown in Figure 8. The reflectivity shows the sharp minimum, and simultaneously the maximum in Raman band intensity at 35.2º (only for the optimum gap size with p-polarized light). Both the reflectivity and Raman band intensity changes drastically to both sides of the resonance angle (see Figures. 8-9). In addition, for the fixed incident angle, the reflectivity and Raman band intensity showed the minimum and maximum at a gap size of 2.5 lL/4. Thus, it is proved that the SPP on Pt is excited as described here and that the Raman band is considerably enhanced. The observed enhancement of ca. 300 for t (CuPc) = 3.5 nm is quite large and is similar to those found over the Ag surface. From a theoretical evaluation, much more modest enhancements around 40 were predicted for randomly orientated CuPc molecules on the Pt surface and is compatible with a larger intrinsic damping in Pt than is found in Ag. As can be seen from Table 2, the parallel orientation of CuPc to the Pt surface gives the largest enhancement. The parallel orientation of the copper phthalocyanine molecules in the layer was confirmed by observing the depolarization ratio. It decreases to zero with decreasing layer thickness [24,26]. Furthermore, this conclusion was also supported by X-ray diffraction and AFM measurements for the Pt films deposited onto BK-7 glass substrate by the AC sputtering method. This Pt layer consists of (111) terraces with a size near 10 X 10 nm and this allows the CuPc molecules to orient parallel to the surface. In contrast, the enhancement of ca. 25 for evaporated Ni films is in accordance with the theoretically predicted value for random orientated CuPc.
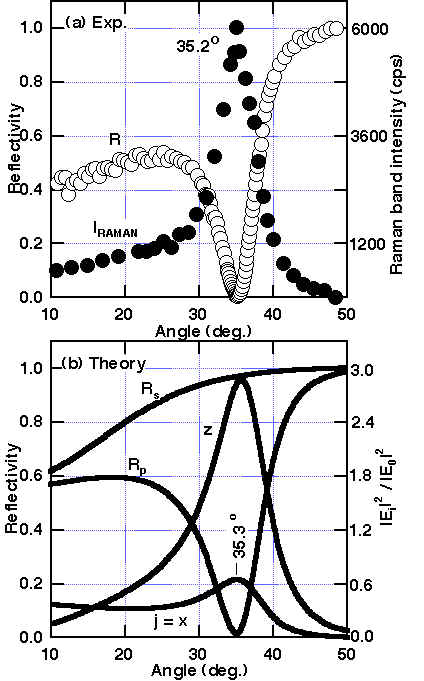
Figure 8. Reflectivity R at p-polarization and unpolarized Raman intensity I from CuPc at 1530 cm-1 in the Weierstrass prism/air gap/CuPc (8.6nm)/platinum system for various incident angles [24]:
(a) observed (R:O, I:•) and (b) theoretical curves, where |Ej|2/|E0|2 is the squared electric field parallel (j=x) or perpendicular (z) to the surface normalized to that of the incident field (j=o). The gap size dopt is 2.5 lL/4 (exp.) or 1.5lL/4 (theory).
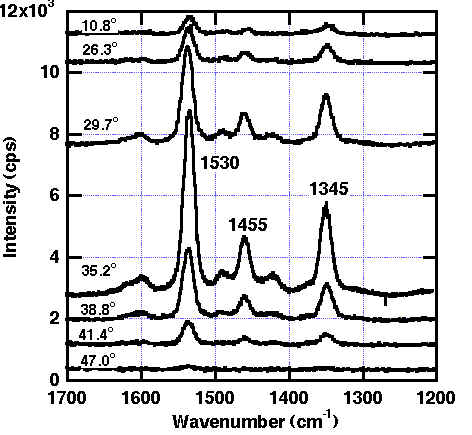 Figure 9. Raman spectra of CuPc on platinum for different incident angles, excited at 5145nm (p-polarized light) [24].
Figure 9. Raman spectra of CuPc on platinum for different incident angles, excited at 5145nm (p-polarized light) [24].
The air gap size is 2.5lL/4
Thus, we have confirmed that ATR-SPP Raman spectroscopy is relevant not only for Au, Ag, Cu but also for the transition metals with higher intrinsic damping. Furthermore, orientation information on adsorbates can be obtained by the comparison of theoretical and experimental enhancement factors.
Application to the electrode surface
Our purpose is to characterize the adsorbates on the electrode surface. Thus, Raman spectra of p-nitrothiophenol [PNTP] – self assembled monolayer [SAM] films on Au and Ag electrode surface were studied under potential control [22]: (a) Raman spectral measurement for the monolayer was confirmed, (b) irreversible electrochemical reduction of PNTP below -0.3 V was elucidated by ATR Raman spectroscopy, (c) the peak potential shifts to positive side with changing the excitation wavelength from 633 to 514nm. This proves the charge transfer from the metal to the lowest unoccupied molecular orbital (LUMO) of p-aminothiophenol (PATP), which is electrochemically reduced from PNTP. Furthermore, Pyridine on smooth Ag surfaces deposited onto a ZnSe prism shows two distinct adsorption states at different potentials [25, 29]. The peak potential of the totally symmetric stretching band of Py at 1004 cm-1 shifts positively by ca. 0.25 V when changing the excitation wavelength from 647.1 nm to 568.2 nm. This result is rationalized by the charge transfer mechanism similar to PATP above. It was also supported by the observation that the roughening procedure of the Ag surface gives only one band at ca. 1009 cm-1. From these results, the charge transfer between the metals and molecular orbital of the adsorbates must occur even for smooth metal surfaces for Py. The other band at 1014 cm-1 shows its peak intensity at a potential of -1.2 V, which is independent of the excitation wavelength. From cyclic voltammetry and ATR-IR measurements, this peak potential corresponds to the maximum surface coverage of Py [29]. Accordingly, we have demonstrated that details on adsorption/desorption phenomena at smooth metal surfaces (and even at the single crystal surface) can be characterized with this method.
Combination of SNOM with the highly-sensitive ATR-Raman spectroscopy
The highly-sensitive ATR-SPP method is useful not only to save time in measurement but also to improve spatial resolution. Spatial resolution beyond the diffraction limit in SNOM results from the fact that the localized evanescent wave is formed in the vicinity of a microscopic aperture and this is much smaller than the wavelength of light [30]. At present, spatial resolution can be achieved ca. 10 nm in SNOM imaging [30]. In addition, fluorescence or Raman spectra have been reported with spatial resolutions of ca. 100 nm i.e., much better than that typical in conventional optical microscopy [31]. We briefly describe below the ATR-SNOM-Raman system developed in our laboratory [32, 33].
The sample surface deposited on the prism is irradiated with the laser beam under the ATR conditions (see Figure 10). The evanescent wave is formed just outside the prism and perturbed by the local optical properties of the sample. The tip of the fibre probe is sharpened down to several tens of nm in diameter by a chemical etching procedure [30]. The distance between the fibre and the sample is controlled using a shear force mode. For details the reader should refer to references [32, 33]. When the fibre approaches the sample surface, the evanescent wave interacts with the electric dipole of the probe to produce propagating light (a SNOM signal). The signal is then transferred in to the fibre and so to the photo multiplier or charge coupled detector via a polychromator to obtain Raman spectra. By this procedure, the highly-sensitive ATR-SPP Raman spectroscopy method becomes particularly useful in detecting extremely weak signals from narrow sample regions. Compared to the illumination mode, this approach has several advantages viz. (a) any fluorescence or Raman signal from the fibre itself does not contribute to the observed spectra; (b) stronger incident laser power is available, because no metal coating is necessary on the fibre; (c) the SPP can be used through this optical configuration to increase the signal intensity. We have confirmed that the SNOM image of sputtered gold films can be obtained with a spatial resolution of several tens of nm, simultaneously with a topographic image as shown in Figure 11. The Raman band from CuPc on the prism was observed at ca. 1530 cm-1 without using any resonance effect, as shown in Figure 12. Furthermore, we have confirmed that the silver island film evaporated underneath the CuPc gives a prominent SERS signal with an enhancement of ca. 100.
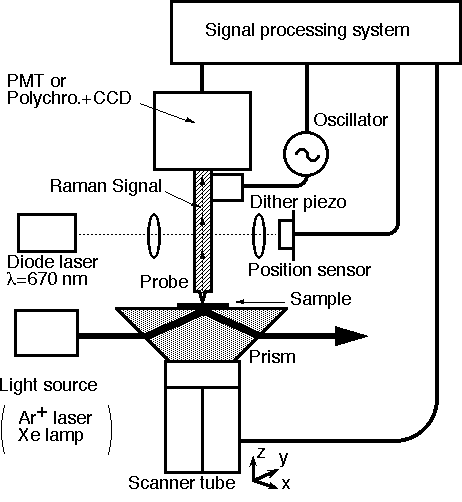
Figure 10. Optical set-up for ATR-SNOM Raman spectroscopy.

Figure 11. Topographical image (a) and SNOM image (b) for the gold films obtained with the device shown in Figure 10.
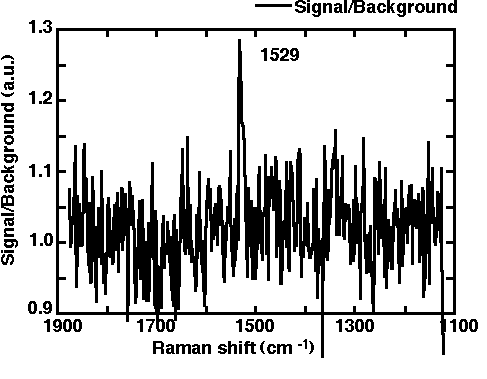
Figure 12. Raman spectra from CuPc on the prism obtained with the equipment shown in Figure 10.
When ultimately molecular level spatial resolution is achieved using the ATR-SNOM-Raman spectroscopy, the method will directly give Raman spectra from individual molecules at different adsorption sites. Clearly, this will be invaluable in elucidating and controlling the electrochemical reaction processes.
Acknowledgment
The author appreciates Prof. Andreas Otto (Heinrich-Heine-Universitaet Duesseldorf, Germany) for useful discussions to develop ATR-SPP Raman spectroscopy.
References
- K. Itaya, Prog. Surf. Sci. 58, 121 (1998).
- J. Lipkowski and P. N. Loss (Eds.) Adsorption of Molecules at Metal Electrodes (VCH, 1992).
- A. Otto, I. Mrozek, H. Grabhorn and W. Akemann, J. Phys.: Condens. Matter., 4, 1143 (1992)
- M. Kerekr (ed.), Surface Enhanced Raman Scattering (SPIE, Vol. MS10, 1990).
- T. M. Cotton, in Spectroscopy of Surfaces, R. H. Clark and R. E. Hester (Eds.), John Wiley & Sons, 1988.
- C. G. Blatchford, J. R. Cambell and J. A. Creighton, Surf. Sci., 120, 435 (1982).
- J. T. Krug, G. D. Wang, S. R. Emory and S. Nie; J. Am. Chem. Soc., 121, 9208 (1999).
- J. E. Demuth, K. Christmann and P. N. Sandra, Chem. Phys. Lett., 76, 201 (1980).
- K. Kneipp, H. Kneipp, I. Itakan, R. R. Dasari and M. S. Feld; Chem. Rev., 99, 2957 (1999).
- M. Futamata, Surf. Sci. 427/428 (1999) 179, Vibrational Spectrosc., 19, 187(1999); Chem. Phys. Lett., 317, 304 (2000).
- A. Otto; Polaritons, E. Burstein and F. de Martini, Eds. (Pergamon Press, New York, 1974) p. 117
- K. Raether, Surface Plasmons (Springer-Verlag, 1988).
- M. Futamata; Langmuir, 11, 3894 (1995).
- L. G. Faegerstam, J. Mol. Recog., 3, 208 (1990).
- Y. Okamoto and I. Yamaguchi, Opt. Commun., 93, 265 (1992).
- S. Ushioda and Y. Sasaki: Phys. Rev., B27, 1401 (1983).
- K. Kurosawa, R. M. Pierce and S. Ushioda: Phys. Rev., B33, 789 (1986).
- J. Giergiel, C. E. Reed,J. C. Hemminger and S. Ushioda: J. Phys. Chem., 92, 5357 (1988).
- C. E. Reed, J. Giergiel, J. C. Hemminger and S. Ushioda: Phys. Rev., B36, 4990 (1987).
- B. Pettinger, A. Tadjeddine and D. M. Kolb: Chem. Phys. Lett., 66, 544 (1979).
- M. Futamata, P. Borthen, J. Thomassen, D. Schumacher and A. Otto; Appl. Spectrosc., 48, 252 (1994).
- M. Futamata; J. Phys. Chem., 99, 11901 (1995).
- M. Futamata; Appl. Opt., 36, 364 (1997).
- M. Futamata, E. Keim, A. Bruckbauer, D. Schumacher and A. Otto; Appl. Surf. Sci. 100/101, 60 (1996).
- M. Futamata; Surf. Sci., 386, 89 (1997).
- M. Futamata, A. Bruckbauer and A. Otto; in preparation.
- W. Wittke, A. Hatta and A. Otto, Appl. Phys. A48, 289 (1989).
- H. D. Abru(a (Ed.), Electrochemical Interfaces (VCH, NY 1991). Chap. 7.
- M. Futamata and D. Diesing; in preparation.
- M. Ohtsu (Ed.), Near-Field Nano/Atom Optics and Technology, (Springer, 1998) Chap. 1.
- For example, D. A. Smith, S. Webster, M. Ayad, S. D. Evans, D. Fogherty and D. Batchelder; Ultramicroscopy, 61, 47 (1995) and V. Deckert, D. Zeisel and R. Zenobi; Anal. Chem. 70, 2646 (1998).
- S. Takahashi, M. Futamata and I. Kojima, Buneski, 47, 1055 (1998) and J. Microsc. 194, 519 (1999).
- M. Futamata and A. Bruckbauer, in preparation.
- Y. Maruyama, M. Ishikawa and M. Futamata, submitted.
- In-coupling denotes the excitation of the SPP with light at the resonance angle
ai=aSPP, where the energy and wavevector of the light (evanescent wave) accord with those for the SPP (see Figure 1). The excited SPP with slightly different energy and wavevector due to Raman scattering from adsorbates is still located on the right side of the light. Thus, the SPP must be transformed into the propagating light to detect, which is achieved by using a prism as a reciprocal process of the in-coupling. This process is called the out-coupling. Although the in- and out-coupling are also realized by scattering of light at roughened metal surfaces, the phenomenon yields another difficulty in discussing the adsorbed state quantitatively (see text).
REF: M. Futamata, Int. J. Vib. Spect., [www.irdg.org/ijvs] 4, 2, 9 (2000)