The Raman Spectrum of Iodine Azide IN3
CONTRIBUTED ARTICLE
Received 23.50 European Standard Time 31-12-99,
accepted: subject to minor corrections 0.21 GMT, 1-1-2000
3. The Raman Spectrum of Iodine Azide IN3
Margaret-Jane Crawford and Thomas M. Klapötke*
Institute of Inorganic Chemistry,
University of Munich (LMU),
Butenandtstraße 5 – 13 (Haus D),
D-81377 Munich (Germany)
fax: +49-89-2180-7492,
e-mail:tmk [at] cup.uni-muenchen.de (tmk@cup. uni-muenchen.de)
The Raman spectra of pure iodine azide in the solid state and in CH2Cl2 solution were recorded at 0°C. Assignment of the vibrations was made on the basis of correlated ab initio computations.
Keywords
Raman, Iodine azide, vibrational analysis.
Introduction
The chemistry of covalent inorganic azides originated with the synthesis of aqueous HN3 solutions by Tony Curtius in 1890.[1] A little later, and exactly 100 years ago, in 1900, it proved possible to prepare iodine azide, as the first member of the now complete series of halogen azides.[2] Over the years the structure of iodine azide has been determined experimentally by microwave spectroscopy,[3] electron diffraction [4] and X-ray crystallography.[5] Spectroscopically IN3 has been characterized by solution 14N NMR [6] and gas-phase IR spectroscopy.[7] The recent resurgence of iodine azide chemistry [8-10] has also stimulated theoretical work on this molecular trans-bent (Cs symmetry) compound (Figure 1).[7,11] For example, the vibrational spectrum of IN3has been calculated by quantum chemical methods.[7,11] However, there is still a considerable degree of confusion concerning the assignment of the vibrational modes in the Raman spectrum of IN3 [7] and the only report of which to our knowledge dates back to 1978.[12] In this contribution we wish to report on the redetermination of the Raman spectrum of IN3 both in the solid state and in CH2Cl2 solution at 0°C using a Nd-YAG IR laser and the assignment of the Raman peaks on the basis of correlated ab initio computations.
![]()
Figure 1. Molecular structure of iodine azide, IN3.
Experimental Section
Caution: Neat IN3 is explosive! The explosive nature increases with greater purity. Appropriate safety precautions should be taken.
Raman spectra: Raman spectra were recorded on a Perkin Elmer 2000 R Raman spectrometer using the 1064 nm exciting line of a Nd-YAG IR laser and the 180° geometry. All spectra were recorded at 0°C using a Ventacon low-temperature cell. Thin-walled 10-mm Pyrex glass tubes were used as sample containers. The solid state spectra were recorded at 50 mW (300 scans) whereas the solution spectra were recorded at 400 mW laser power (100 scans).
Preparation of iodine azide, IN3: IN3 was prepared by the reaction of an excess of dry, freshly prepared silver azide, AgN3 (from AgNO3 and NaN3, both Aldrich), and iodine (Aldrich) in CFCl3 (Merck) solution.
At 0°C 1.20 g (8.0 mmol) of AgN3 were suspended (magnetic bar) in a PTFE beaker in 20 mL of CFCl3, and then 0.5 g (2.0 mmol) of I2 were added to the stirred suspension. The reaction mixture was stirred at 0°C and allowed to react for 1 h. Since both AgN3(excess) and AgI are insoluble in CFCl3, the clear yellow IN3/CFCl3 solution was poured into two 10-mm glass tubes (ca. 10 mL into each tube). The 10-mm glass tubes were kept at 0°C, and the solvent (CFCl3) was pumped off at 0°C and 30 Torr (membrane vacuum pump).
Raman spectra on solid IN3 were recorded directly using the 10-mm sample tube containing ca. 1.0 mmol (169 mg) IN3. For recording the solution spectra, solid IN3 (1.0 mmol, 169 mg) was redissolved in 1 mL CH2Cl2 (c = 1.0 mol L-1).
Computational Methods: The structure, total energy (E), vibrational frequencies and zero point energies (zpe) of IN3 were computed ab initio and fully optimized at the HF and electron correlated MP2(FC), MP2(FULL) and QCISD levels of theory with the program package Gaussian 98.13 In addition density functional hybrid computations at the B3LYP level of theory were performed. For a concise definition of the MP2, B3LYP and CI methods see refs. [14, 15]. For one set of calculations for N a D95 Dunning/Huzinaga full double zeta basis set was used;16 for I a quasi-relativistic pseudopotential (LANL2DZ, Los Alamos potential) 17-19 was used where the basis functions for the valence s and p electrons consist of the standard double-z basis set (notation HF/LANL2DZ, MP2/LANL2DZ etc.). For the other set of calculations for N Dunning’s correlation consistent cc-pVTZ triple-z basis set was used (4s,3p,2d,1f);20-24 and for I a quasi-relativistic pseudopotential (I-ECP-MDF 4 46, Stuttgart ecp) 25 was used where the basis functions for the valence s and p electrons are treated with a (7s7p2d1f)/[3s3p2d1f] valence basis set (notation HF/MDF, MP2/MDF etc.).25
Results and Discussion
Figure 2 shows the Raman spectrum recorded on neat IN3, and Figure 3 shows the Raman spectrum recorded on a solution (1 mol L-1) of IN3 in CH2Cl2 (see experimental).
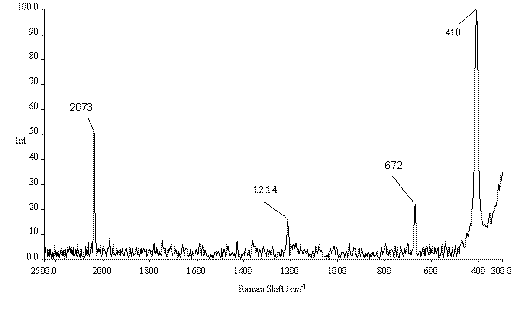
Figure 2. Raman spectrum of solid iodine azide, IN3 (0°C).
In the solid state the Raman spectrum of IN3 shows four characteristic peaks at 2073, 1214, 672 and 410 cm-1 which can be assigned to the n 6 (asymmetric N3 stretching mode), n 5 (symmetric N3 stretching mode), n 4 (d -NNN) and n 2 (N-I stretch) modes of IN3 (cf. Table 1). In CH2Cl2 solution two of these vibrations are significantly shifted to 2056 and 662 cm-1, which can be ascribed to the transition from the polymeric structure in the solid state to the molecular appearance of IN3 in solution. Whereas in solution the symmetric azide stretch (n 5) was not observed the N-I stretch (n 2) remained essentially unchanged at 411 cm-1. In addition, the n 3 peak at 535 cm-1 was observed in solution but not in the solid state.
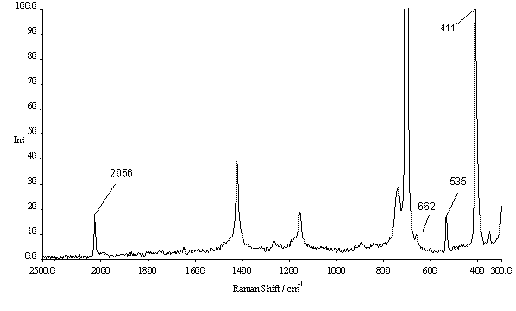
Figure 3. Raman spectrum of iodine azide, IN3, dissolved in CH2Cl2 (0°C, 1 mol L-1); the unlabelled peaks all originate from the CH2Cl2 solvent.
It is interesting that the symmetric mode n 5 (n s-NNN) was only observed in the solid state but not in solution although symmetric modes are conventionally strong in Raman and weak in IR. This, however, is in accord with the earlier Raman study in which the asymmetric mode (n 6) was observed to be of medium intensity whereas the symmetric mode (n 5) was only observed as a weak band.12
| HF/LANL2DZ | MP2(FC)/LANL2DZ | MP2(FU)/LANL2DZ | QCISQ/LANL2DZ | B3LYP/LANL2DZ | exptl. c | |
| -E / a.u. | 174.300582 | 174.702889 | 174.705670 | 174.685096 | 175.492974 | |
| el. state | 1A’ | 1A’ | 1A’ | 1A’ | 1A’ | |
| zpe /kcal mol-1 | 7.3 | 7.2 | 7.2 | 6.1 | 6.8 | |
| d(I-N1)/Å | 2.122 | 2.166 | 2.167 | 2.192 | 2.217 | 2.12(1) |
| d(N1-N2)/Å | 1.274 | 1.315 | 1.315 | 1.306 | 1.275 | 1.26(1) |
| d(N2-N3)/Å | 1.123 | 1.200 | 1.200 | 1.181 | 1.174 | 1.15(1) |
| <(INN) / ° | 114.4 | 114.3 | 114.2 | 113.1 | 114.4 | 107(1) |
| <(NNN) / ° | 173.2 | 168.7 | 168.8 | 189.8 | 171.2 | 170(3) |
| n 1 | 185 (14) | 147 (12) | 146 (12) | 158 | 162 | n.o. |
| n 2 | 415 (94) | 372 (14) | 372 (14) | 338 | 357 | 411 (10) |
| n 3 | 547 (2) | 407 (3) | 408 (3) | 432 | 483 | 535 (2) |
| n 4 | 656 (31) | 621 (3) | 621 (3) | 564 | 614 | 662 (1) |
| n 5 | 1094 (60) | 1072 (12) | 1073 (12) | 1008 | 1147 | n.o. d |
| n 6 | 2214 (256) | 2396 (29) | 2395 (30) | 1801 | 1984 | 2056 (3) |
Table 1 Computational a and experimental results for IN3 b
a For N a D95 Dunning/Huzinaga full double zeta basis set was used;16 for I a quasi-relativistic pseudopotential (LANL2DZ, Los Alamos potential).
b rel. Raman intensities in Å4 amu-1
c structural data from gas-phase electron diffraction [ref. 4], CH2Cl2 solution Raman data (see text); n.o. = not observed
d The n 5 band was only observed in the solid state at 1214 cm-1.
| HF/MDF | MP2(FC)/MDF | B3LYP/MDF | MP2 scaled by 0.93 13b | B3LYP scaled by 0.955 13b | exptl. c | |
| -E / a.u. | 174.487478 | 175.227655 | 175.651634 | |||
| el. state | 1A’ | 1A’ | 1A’ | 1A’ | 1A’ | |
| zpe /kcal mol-1 | 8.4 | 7.6 | ||||
| d(I-N1)/Å | 2.033 | 2.055 | 2.081 | 2.12(1) | ||
| d(N1-N2)/Å | 1.230 | 1.244 | 1.234 | 1.26(1) | ||
| d(N2-N3)/Å | 1.089 | 1.152 | 1.130 | 1.15(1) | ||
| <(INN) / ° | 112.5 | 111.6 | 113.1 | 107(1) | ||
| <(NNN) / ° | 174.9 | 172.4 | 172.6 | 170(3) | ||
| n 1 | 200 (8) | 165 (8) | 175 | 153 | 167 | n.o. |
| n 2 | 495 (43) | 461 (17) | 421 | 428 | 402 | 411 (10) |
| n 3 | 676 (1) | 537 (1) | 572 | 500 | 546 | 535 (2) |
| n 4 | 777 (20) | 699 (4) | 683 | 650 | 652 | 662 (1) |
| n 5 | 1284 (13) | 1232 (10) | 1255 | 1146 | 1198 | n.o. d |
| n 6 | 2449 (198) | 2257 (16) | 2185 | 2099 | 2087 | 2056 (3) |
Table 2 Computational a and experimental results for IN3 b
a For N Dunning’s correlation consistent cc-pVTZ triple-z basis set was used (4s,3p,2d,1f);20-24 and for I a quasi-relativistic pseudopotential (I- ECP-MDF 4 46, Stuttgart ecp) 25 was used where the basis functions for the valence s and p electrons are treated with a (7s7p2d1f)/[3s3p2d1f] valence basis set (notation HF/MDF, MP2/MDF etc.).25
b rel. Raman intensities in Å4 amu-1
c structural data from gas-phase electron diffraction [ref. 4], CH2Cl2 solution Raman data (see text); n.o. = not observed
d The n 5 band was only observed in the solid state at 1214 cm-1.
Whereas IN3 has strong intermolecular interactions in the solid state, it is assumed to be monomeric even in relatively concentrated solutions.[6] Therefore it can be expected that the observed vibrational frequencies for an IN3 solution sample in a non-polar solvent (CH2Cl2) are comparable to those computed for an isolated IN3 molecule. The relatively poor agreement between experiment and theory even at high levels of theory (MP2(FULL) and QCICD) when for N a D95 Dunning/Huzinaga full double zeta basis set was used;[16] and for I a quasi-relativistic pseudopotential (LANL2DZ, Los Alamos potential) was applied (Table 1) indicates that the basis set was too small and especially polarization was not accounted for enough. Table 2 on the other hand shows that the agreement between experiment and theory is sufficient to good at electron correlated MP2 and B3LYP level when for N the far more expensive Dunning’s correlation consistent cc-pVTZ triple-z basis set was used [4s,3p,2d,1f];[20-24] and for I a quasi-relativistic pseudopotential (I-ECP-MDF 4 46, Stuttgart ecp) [25] where the basis functions for the valence s and p electrons are treated with a (7s7p2d1f)/[3s3p2d1f] valence basis set (notation HF/MDF, MP2/MDF etc.).[25] In addition, it is noteworthy that also the computed relatively expensive MP2 Raman intensities fit very well with the experimentally obtained data. Thus, the question as to whether the Raman band at 411 cm-1 originates from the N-I stretch in IN3 [7,11,12] can finally be answered with confidence.
These results clearly show that for the prediction of the vibrational frequencies for heavy elements such as iodine it is important not only to sufficiently account for electron correlation but also to use rather large polarized basis sets, at least for the valence electrons when applying an effective core potential.
Conclusions
The Raman spectra of iodine azide, IN3, were recorded for neat IN3 and a solution of IN3 in CH2Cl2. Assignment of the spectra was made on the basis of correlated high-level ab initio computations.
Acknowledgment
The authors wish to thank the University of Munich, the Fonds der Chemischen Industrie and the Deutsche Forschungsgemeinschaft (KL 636/6-1) for financial support.
References
- N. N. Greenwood, A. Earnshaw, Chemistry of the Elements, Pergamon, Oxford, New York, 1984.
- A. Hantzsch, M. Schümann, Ber. Dtsch. Chem. Ges. 1900, 33, 522.
- H.-O. Munz, H.-K. Bodenseh, T. M. Klapötke, 14th Colloquium on High Resolution Molecular Spectroscopy, September 11 – 15, 1995, Dijon.
- M. Hargittai, J. Molnar, T. M. Klapötke, I. C. Tornieporth-Oetting, M. Kolonits, I. Hargittai, J. Phys. Chem. 1994, 98, 10095.
- P. Buzek, T. M. Klapötke, P. v. R. Schleyer, I. C. Tornieporth-Oetting, P. S. White, Angew. Chem. 1993, 105, 289; Angew. Chem. Int. Ed. Engl. 1993, 32, 275.
- P. Geißler, T. M. Klapötke, H.-J. Kroth, Spectrochim. Acta, Part A 1995, 51A, 1075.
- A. Schulz, I. C. Tornieporth-Oetting, T. M. Klapötke, Inorg. Chem. 1995, 34, 4343.
- I. C. Tornieporth-Oetting, T. M. Klapötke, Angew. Chem. 1995, 107, 509; Angew. Chem. Int. Ed. Engl. 1995, 34, 511.
- T. M. Klapötke, Chem. Ber. 1997, 130, 443.
- Covalent Inorganic Non-Metal Azides, I. C. Tornieporth-Oetting, T. M. Klapötke, in: Combustion Efficiency and Air Quality, I. Hargittai, T. Vidoczy (Eds.), Plenum Press, New York, 1995, p. 51.
- M. Otto, S. Lotz, G. Frenking, Inorg. Chem. 1992, 31, 3647.
- U. Engelhardt, M. Feuerhahn, R. Minkwitz, Z. Anorg. Allg. Chem. 1978, 440, 210.
- a) Gaussian 98, Revision A.3, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head- Gordon, E. S. Replogle, and J. A. Pople, Gaussian, Inc., Pittsburgh PA, 1998;b) A. Frisch, M. J. Frisch, Gaussian 98 User’s Reference, Gaussian, Pittsburgh, 1998, and references cited therein.
- W. J. Hehre, L. Radom, P. v. R. Schleyer, J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, 1986.
- T. M. Klapötke, A. Schulz, R. D. Harcourt, Quantum Chemical Methids in Main-Group Chemistry, Wiley, Chichester, 1998;
- T. H. Dunning, Jr., P. J. Hay, in Modern Theoretical Chemistry, H. F.Schaefer, III (ed.), Plenum, New York, 1976, vol. 3, p. 1.
- P. J. Hay, W. R. Wadt, J. Chem. Phys. 1985, 82, 270.
- W. R. Wadt, P. J. Hay, J. Chem. Phys. 1985, 82, 284.
- P. J. Hay, W. R. Wadt, J. Chem. Phys. 1985, 82, 299.
- D. E. Woon, T. H. Dunning, Jr., J. Chem. Phys. 1993, 98, 1358.
- R. A. Kendall, T. H. Dunning, Jr., R. J. Harrison, J. Chem. Phys. 1992, 96, 6796.
- T. H. Dunning, Jr., J. Chem. Phys. 1989, 90, 1007.
- K. A. Peterson, D. E. Woon, T. H. Dunning, Jr., J. Chem. Phys. 1994, 100, 7410.
- A. Wilson, T. v. Mourik, T. H. Dunning, J. Mol. Struct. 1997, 388, 339.
- M. Dolg, to be published.
REF: M-J Crawford & T.M. Klapötke Int. J. Vib. Spect., [www.irdg.org/ijvs] 3, 6, 3 (1999/2000)