Raman and SERS spectroscopy for probing drug-target interactions: from in-vitro models to intracellular imaging
Ganesh D Sockalingum,
Serguei Charonov,
Abdelilah Beljebbar,
Hamid Morjani,
Michel Manfait & Igor Chourpa*
Unité MéDIAN, IFR 53, UFR de Pharmacie,
Université de Reims Champagne-Ardenne,
51 rue Cognacq-Jay,
51096 Reims cedex,
France
* Laboratoire de Chimie Analytique,
Faculté des Sciences Pharmaceutiques,
Université de Tours,
37200 Tours,
France
Tel: (33) 3 26 91 35 74 or (33) 3 26 91 35 53
e-mail to: ganesh.sockalingum [at] univ-reims.fr
or to:michel.manfait [at] univ-reims.fr
Abstract
In this work we describe the SERS approach with a view to investigating the interactions between drugs and their pharmacological targets. Two different drugs which have different cellular targets are considered. In both cases spectral features of drug-target complexes in-vitro and at the cellular-level are compared. The first example deals with FT-Raman and -SERS measurements of a retinoid analogue, dimethylcrocetin, a natural product which complexes to the human retinoic acid receptor, present in certain cancer cell lines only. In the second case, we show SERS data of an antitumor drug mitoxantrone, collected from a single live cell. This drug has DNA as its target and comparison is made with in-vitro experiments. Finally, we show how SERS data of mitoxantrone obtained at the single cell level are used for spectral imaging.
Keywords
Anti-tumour drugs, Live cancer cells, Raman spectroscopy, SERS, Spectral imaging
Introduction
Raman spectroscopy, in its simplest forms, is not a sensitive spectroscopic technique since Raman scattering cross-sections for most molecules are very small whereas the extinction coefficients in optical absorption or specific emissivities in emission spectroscopy. Fluorescence from the molecule of interest or other sample components further compromises detection, as the luminescence background often obscures a weak scattering signal. The investigator working at the level of the living cell is severely handicapped by these limitations since on the one hand cell components fluoresce strongly and on the other Raman sensitivity is seldom not enough to detect drugs at physiological concentrations. However, when these barriers to the use of Raman spectroscopy can be overcome, the method becomes a powerful tool for biological and biomedical research owing to its capacity to give molecularly specific and structurally useful information from such samples in a non-destructive and non-invasive manner. It is also one of the few available techniques that allow the selective study of molecular interactions within high-molecular-weight supramolecular complexes. The problem of sensitivity can be addressed either by using resonance Raman (RR), surface-enhanced Raman spectroscopy (SERS) or a combination of both (SERRS). Early studies have shown that RR is very useful because of its selectivity to certain chromophores and sensitivity to monitor drug-DNA complexes [1, 2]. But these advantages are exploitable mainly in vitro at relatively high concentrations and exist in the absence of fluorescence interference. For physiological concentrations of drugs (10-6 M or less ), RR is not always sufficient, and also not all drugs possess a resonant moiety. SERS is a technique in which lasers are used to excite vibrational transitions in molecules adsorbed on a rough metallic substrate [3, 4]. As a result of large optical fields and resonance-related effects, the Raman cross-section of a molecule on a surface is enhanced by factors of up to 106. When the SERS effect is coupled with an optical resonance in the molecule (SERRS), even larger enhancement can be observed. SERS is quite a good alternative because the process allows us to solve both sensitivity and fluorescence (quenching) problems. Another potential solution to the problem of fluorescence is NIR Fourier-transform (FT) Raman spectroscopy. This technique is less sensitive but offers the advantage of being rapid and causes less sample damage. Combined with the SERS technique, it can be an attractive method for probing the cell. To have access to molecular level information on drug-target interactions, both in-vitro and at the cellular level, our efforts have been mainly directed to the use of SERS, FT-Raman spectroscopy and combinations of both. The question is how can these techniques be adapted to solve specific problems such as drug-target interactions at the cellular level? This issue is important because understanding such molecular interactions help to have a better insight into the mechanisms of action and hence improve the development of more effective drugs.
Thus, there are several considerations to be taken into account prior to the application of SERS to such biological studies. It is well known that the SERS effect exhibits certain properties that distinguish it clearly from the normal Raman effect. First, the Raman cross-sections for adsorbed molecules are generally a factor of 103-106 larger than those of non-adsorbed ones. Second, the SERS spectrum may differ from that of the non-adsorbed free molecule in the sense that selective enhancement of certain bands or appearance of new bands may occur, often due to the contribution of the chemical component of the SERS effect. Finally, studies at cellular level implies the use of a micro-configuration. When SERS is coupled to a microscope one has in hand a powerful microprobe. This idea was first presented by Van Duyne et al. [5], who calculated detection limits of less than 1 amol, corresponding to about 105 (SERS) and ca. 600 (SERRS) molecules in the probe beam of their experiments.
Although electrodes are often the substrates of first source for SERS substrates investigations of most biological samples, but they are not ideal. This is mainly because biological systems are more complex, SERS-active electrode surfaces are quite tedious to prepare, and are not stable although they provide a good sensitivity [6]. Thus, other substrates such as colloids and vacuum-deposited metal island films have been developed and tested as a response to the problemsarising from biological systems [7-10]. Both colloids and island films exhibit the necessary morphological features that are essential for the enhancement of the Raman cross-section of the adsorbed molecule. Apart from the type and morphology of the metallic surface, which must be rough and must have specific adsorption properties [11], the enhancement of the Raman signal is largely dependent on the excitation frequency [12] and, the chemical nature of the adsorbate. In general, silver colloids provide a reasonable enhancement from the visible to the NIR, silver island films are more suited for the visible, while gold island films and gold colloids work best from the red to the NIR [13].
Chemically prepared colloidal suspensions provide a certain number of advantages for SERS observation, including ease of formation and manipulation, simple characterisation by absorptiometric techniques and a definite dependence of the enhancement on particle size and shape. One major disadvantage is that the SERS activity of a given analyte depends to a certain extent on the method of sol preparation and on the exact protocol used for a given preparation. While the same problem also exists for metal island films, these provide perhaps the most reproducible substrate in terms of enhancement factors. However, since these are static substrates, there is always a risk that the buffer, solvent, or the intense laser fields associated with SERS, destroys the surface morphology that gives rise to surface enhancement.
In-vitro and cellular DMCRT-Rarg interactions:
Raman and SERS measurements
Here, we give an example of how Raman spectroscopy can be used to selectively probe the interactions when dimethylcrocetin forms complexes with the human retinoic acid receptor RARg . In Figure 1 are displayed the RR (457.9 nm) and micro-FT-SERS (1064 nm) spectra from silver colloids of free DMCRT. Comparison of these spectra show that there are hardly any differences in the main band positions and relative intensities. This is an important observation since it tells us that (a) the adsorbed molecule, the analyte is not perturbed by its interaction with the colloidal particles and (b) spectral modifications observed upon complexation could then be reliably interpreted as a consequence of the drug-target interactions. To monitor the interactions in the DMCRT-RARg complex, we will pay particular attention to the bands positioned around 1540, 1165 and 1210 cm-1 assigned respectively to u (C=C) , (indicative of the degree of conjugation), and u (C-C) motions (inform on the isomerisation state). When the micro-FT-SERS spectra of free DMCRT and those of DMCRT-RARg are compared (see Figure 2), the following differences can be noticed upon complexation: (a) there is a global loss of spectral intensity for the complex, (b) the I1541/I1165 ratio remains constant, (c) the I1541/I1210 decreases, and (d) the band at 1210 cm-1 is upshifted. Following these observations that can be correlated to the interactions in the complex only, we can now see what happens at the cellular level. Two type of cell lines have been tested: K562, a human leukemic cell line established from a patient with chronic myelogeneous leukaemia in blast transformation and HL60, a human cell line established from the peripheral blood leukocytes of a patient with acute promyelocytic leukaemia. It has been shown that other retinoids such as all-trans retinoic acid induce differentiation of HL60 but not of K562 cells [14, 15]. This response is supposed to be due to the presence of the specific retinoid binding protein also called RAR, present in the HL60 cell nucleus but not in K562. This interaction leads to the formation of a retinoid-RAR-DNA ternary complex and subsequently specific gene expression [16]. The HL60 and K562 cell lines were incubated in the presence of 5 µM of DMCRT for 4 hours and washed twice with 0.9 % NaCl. After centrifugation, the pellets were then deposited on glass slides and left for 20 min at 37° C before being analysed by FT-Raman microspectroscopy. Due to the accumulation of the drug in the cells and the strong signal from the retinoid, it was possible using FT-Raman to record the spectra directly from the pellets. The cellular level data are compared to those of the free drug in Figure 3. It can be clearly seen that the spectrum obtained from the K562 cell corroborates with that of the free drug and that obtained from the HL60 cell can be correlated with that of the in-vitro experiment, with the DMCRT-RARg complex. When all spectral data are compiled in Table I and compared, the same four remarks observed for the in-vitro complex also apply to the HL60 cell experiment.
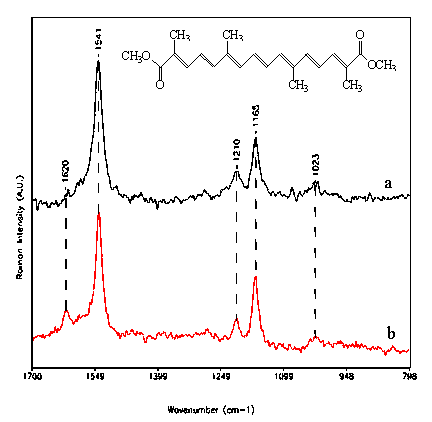
Figure 1. In-vitro spectra of dimethylcrocetin
a: Resonance Raman (457.9 nm) at 5×10-4 M.
b: Micro-FT-SERS (1064 nm) at 5×10-6 M.
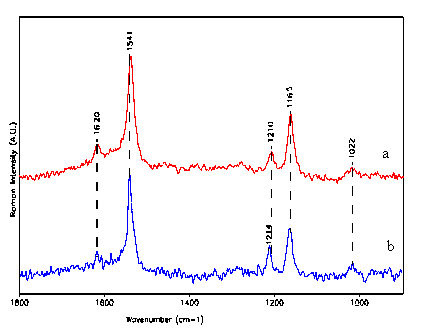 Figure 2. In-vitro Micro-FT-SERS spectra of dimethylcrocetin :
Figure 2. In-vitro Micro-FT-SERS spectra of dimethylcrocetin :
a: free form at 5×10-6 M
b: DMCRT-RARg complex at concentration of 10-5 M
(a) I1541/I1165 = 1.58 I1541/I1210 = 2.5
(b) I1541/I1165 = 1.58 I1541/I1210 = 1.9
Note: Dimethylcrocetin can interact with the human nuclear receptor RAR-g
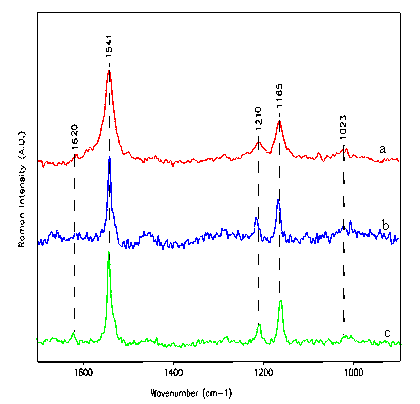
Figure 3. In-vitro and cellular spectra of dimethylcrocetin
(a) in solution, (b) in HL60 cell and (c) in K562 cell
(a) I1541/I1165 = 1.6 I1541/I1210 = 2.5
(b) I1541/I1165 = 1.5 I1541/I1210 = 1.8
(c) I1541/I1165 = 1.7 I1541/I1210 = 2.6
a andt c are similar : absence of any interaction between DMCRT and cell receptor. a and b are different : DMCRT interacts with the cell receptor.
| RR | FT-SERS
(silver colloids) |
FT-Raman | |||||
| Intensity Ratio | Free DMCRT
(5×10-4 M) in-vitro |
Free DMCRT
(5×10-6 M) in-vitro |
DMCRT-RAR
in-vitro |
DMCRT
in HL60 cells |
DMCRT
in K562 cells |
||
| I1541/I1165 | 1.6 | 1.58 | 1.58 | 1.5 | 1.7 | ||
| I1541/I1210
I1165/ I1210 |
2.5
1.56 |
2.5
1.58 |
1.9
1.2 |
1.8
1.2 |
2.6
1.53 |
||
Table 1. I1541/I1165 and I1541/I1210 and I1165/ I1210 Raman signal intensity ratios for RR, FT-SERS and FT-Raman spectra of Dimethylcrocetin in-vitro and at cellular level
From these spectra and table, it can be seen that the I1541/I1165 ratio is almost constant throughout the whole set of experiments. For data obtained from the HL60 cells, the I1541/I1210 ratio is similar to that of the DMCRT-RAR in-vitroexperiment whereas the I1165/ I1210 ratio decreases but has the same value in both sets. Values obtained for the K562 cells correlates with the free DMCRT in-vitro data both for I1541/I1210 and I1165/ I1210 ratios. These data together with the upshift of the 1210 cm-1 band, observed in HL60 and DMCRT-RAR in-vitrocomplex, but not in K562 cells, tell us that there is an interaction, measurable by Raman spectroscopy, between the drug and its cellular target. These results are the outcome of three independent measurements. This study therefore demonstrates that FT-Raman and FT-SERS can be used as powerful probes for investigating the effect of differentiating agents (retinoids and carotenoids) in cancer cells related to the receptor expression.
Now, both K562 and HL60 are leukemic cell lines. Thus, Raman spectroscopy can allow us to distinguish between different cell types and can help to discriminate between sensitive and drug-resistant ones. This was not the aim of this work here. What we have shown is that using two cell lines, one containing a retinoid receptor (HL60) and the other does not (K562, control experiment), Raman spectroscopy can be used to monitor the interaction of the drug with its target. In one case there is a possibility of interaction and in the other there is not. This is observed from the spectral changes. So, the usefulness of the technique here is to (i) predict an interaction, and (ii) to select and improve new retinoids (e.g., synthetic ones) in view of a better interaction with their targets.
Another approach to such studies is to use other types of SERS-active substrates such as gold island films in conjunction with red or NIR excitations and dispersive instruments equipped with CCD detectors. Such systems are very sensitive and appropriate for studies at the cellular level. The absorption frequency maximum of gold island films shifts from the red to the NIR as their mass thickness increases. Although their preparation takes longer in comparison with other substrates, island films offer some control over the morphology and optical properties. This category of SERS substrates could be attractive for applications to biological systems since they can generate a fairly reasonable (but less than with silver colloids) long-range electromagnetic enhancement [13]. This property is important because the signal must originate from the molecule in the vicinity of the metal without being perturbed by the latter.
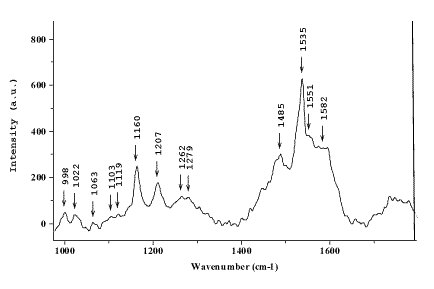
Figure 4. Micro-SERS of a single HL60 cell incubated with 5×10-6 M dimethylcrocetin and placed on a 50 Å thick Au island film. Excitation: 632.8 nm He/Ne laser, 100X objective.
Figure 4 shows the SERS signal collected from a single HL60 cell treated with DMCRT, placed over a 50Å thick gold island film, and excited with a He/Ne laser. By looking at the spectrum, one can recognise the DMCRT bands already discussed although they are slightly shifted. It can also be noticed that not only the signal from the DMCRT is enhanced, but information from the cellular components is also available. For instance, the bands at 999 cm-1(phenylalanine), 1022 and 1063 cm-1 (DNA backbone), 1262 and 1279 cm-1massif (amide III), 1485 and 1582 cm-1 (these may arise from guanine and adenine). This spectrum contains more spectral information than those recorded before but is still dominated by the fluorescence background from the cell. So, the approach using island film SERS substrates (as an area probe external to the cell) for intracellular imaging can be envisaged. However, one has to be careful about the origin of the signal, whether it is intracellular or extracellular, since some drugs can quickly efflux out of the cell. Thus, different parameters have to be controlled before interpreting such spectra.
SERS intracellular imaging of drugs in single live cells
Instrumental considerations
Spectral imaging combines technologies like conventional imaging, spectroscopies and quantitative analysis to produce images for which the spectral signature is associated with each spatial element or pixel. It thus provides the possibility of using different spectral features and not only the integrated intensity within a given spectral range. A major recent development in spectroscopic instrumentation, that has contributed to spectral imaging, is based on the extension of the principles of confocal microscopy (see also article by Otto and Greeve in Int.J.Vib.Spect., [www.ijvs.com] 2, 3, 7 (1998). This mode gives a remarkably improved submicrometric spatial resolution, mostly in the vertical direction [17].
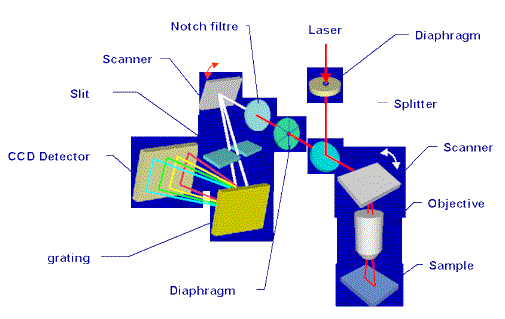
Figure 5. Raman Instrumentation
The scheme of a typical system for spectral imaging is illustrated in Figure 5. To scan a laser beam over a sample, either a motorised XY stage and/or mirror scanners can be used. The illumination schemes that can be applied to generate spatial images can be grouped into four main classes, namely (i) global, (ii) point, (iii) line, and (iv) plane.
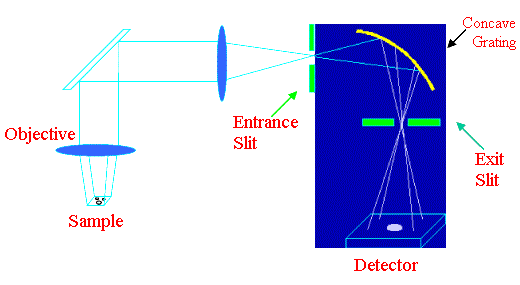
Figure 6. Global illumination scheme
The spectral image in this scheme is generated from band-pass filter images recorded at a successive range of wavelengths. The optical path is the same as for a conventional white light image. The investigated part of the sample is illuminated by the laser beam. The global illumination scheme is now almost never used. This is to indicate that the mode exists. A spectrograph filters the light with a band-pass d n which can be tuned and transmits a monochromatic image onto a 2D-detector. The spectral image is then generated from these filtered images and recorded successively for the whole desired wavelength range. The spectral resolution is weak and recording times for each image are very long. A grating spectrograph is used as a wavelength filter and transmits a monochromatic enlarged image onto a 2 dimensional detector. The position of the band-pass can be displaced by turning the grating [18]. The main disadvantages of this scheme are non-confocality, the low spectral resolution and the very large accumulation times for each image. This scheme is not often used for cell studies.
Figure 7. Point illumination scheme.
Only one sample point is irradiated by the laser and the corresponding spectrum is recorded
The point illumination scheme is the simplest procedure. Only one spatial point of the sample is illuminated by the laser beam and the spectrum corresponding to this point is recorded. Then, either the motorised sample stage moves the sample under the immobile laser focus or the turning mirror changes the position of the laser beam on the sample to the next investigated point. The main disadvantage of the point illumination scheme is the very low speed of spectral acquisition (tens or hundreds of minutes) which is a limitation to its application.
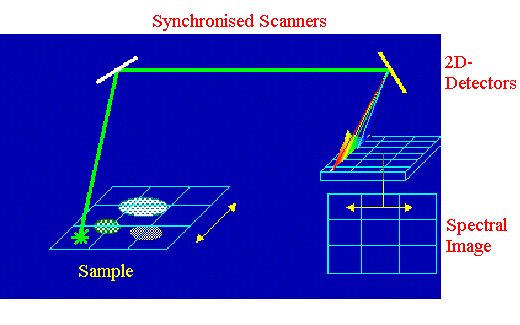
Figure 8. Line illumination scheme
A sample line is irradiated by the laser and the corresponding spectra are recorded
The illumination method is also possible incorporating a two-dimensional detector for simultaneous accumulation of many spectra [19]. One dimension of the detector is used for spectral information contained in the signal and the second one corresponds to the spatial distribution of the signal. The scanning mirror system should consist of two synchronised scanners. The first one deflects the probe laser beam onto the entrance of the microscope objective and produces a narrow line on the sample. The second one deflects the collected signal onto the detector. Scanning of the other space dimension can be achieved by moving the sample in a direction perpendicular to laser line using a motorised stage.
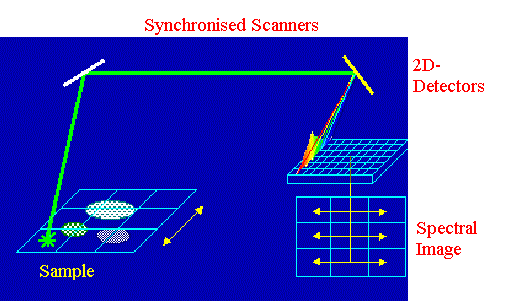
Figure 9. Plane illumination scheme
A rectangular zone of the sample is illuminated by the laser and the corresponding spectra are recorded
Like the line illumination scheme, the plane illumination method makes use of a two-dimensional detector and two synchronised scanners [19]. The scanners must provide the possibility of displacing the probe laser beam in both X and Y space dimensions. It allows us to carry out simultaneous accumulation of spectra from different points on the parallel lines covering the investigated area of the specimen. Displacing the laser beam can be done by turning the scanner along two perpendicular axes. By changing the amplitudes and periods of motion corresponding to these two axes, different space resolutions can be achieved. In this way, the data on the detector are organised as strips of rows. Each strip corresponds to one line on the sample, and each row contains a spectrum originating from one point of this line. The capabilities of the CCD detectors used in modern scanning microspectrometers (~2000 pixels) allow us to accumulate the spectral image ~ 50×50 points. The very important advantage of the plane illumination scheme is the practically simultaneous scanning of the total image, and this significantly reduces the time-caused image distortions in unstable samples.
Intracellular Applications
We have reported recently resonance Raman and SERS studies of antitumor drugs on silver colloids [20-23]. In this section we give an example of how SERS signal of a drug can be collected in the vicinity of colloidal aggregates inside a living cell. This situation is different from that with films, discussed, since now the probe is inside the cell. The drug studied here is mitoxantrone, which is an anthraquinone anticancer drug structurally related to DNA-anthracycline antibiotics [24]. Mitoxantrone is a very potent drug and is used in the treatment of many types of cancer. It is able to cause condensation and compaction of chromatin, to disturb DNA-protein interactions and to trap the cleavable complex between DNA and nucleus-located DNA topoisomerase II. However, before the investigation of mitoxantrone inside a single live cell, we recorded RR and SERS spectra of the drug and its complexes with DNA [see also ref. 21]. The study of drug/target complexes in solution is a necessary prerequisite for the intracellular spectroscopic investigations. In vitro spectral characterisation of the free and complexed drugs is important for a better interpretation of the spectra recorded from cellular compartments.
For mitoxantrone, the experimental conditions used allowed the adsorption of the drug and even of the drug/DNA complexes without detectable perturbations of the drug or of the molecular interactions within the complex (see Figure 10 and comments therein). On the basis of these experiments and on band assignments by comparing RR and SERS, it has been possible to infer a preferential intercalation of rings A and B of the chromophore with the DNA double helix, while ring C remains outside. We have also confirmed this finding by NIR-FT-SERS experiments [25]. In fact, at very far off-resonance conditions of excitation at 1064 nm, CC-N and CC-O deformation modes at 441 and 466 cm-1 and localised on rings A and C of the chromophore respectively, are strongly enhanced compared to the RR and normal SERS spectra. We observed that in the drug-DNA complex, the band at 441 cm-1 disappeared, while that at 466 cm-1 remained the same and was as strong as that near ~1300 cm-1 band. The latter is the strongest feature in RR, pre-RR, SERS and FT-SERS and has been assigned to the ring stretching mode coupled with C-O stretching motions. As has been observed, this band, is very sensitive to the C-O group in ring A of the mitoxantrone molecule, decreases upon complexation.
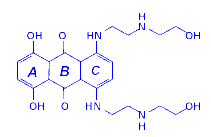
Mitoxantrone (MXT)
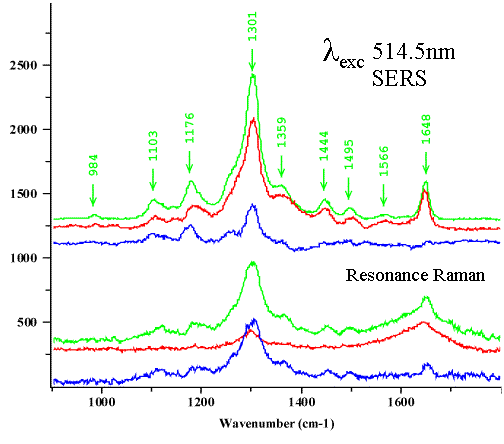
Figure 10. In-vitro models of mitoxantrone
and its complexes with DNA.
I.Adsorption on the silver surface does not induce any detectable perturbations either drug/DNA complex.
II. Preferential DNA intercalation of rings A-B of the chromophore
The next step was to record SERS spectra from a single K562 cell by performing spectral imaging. The procedure is schematically depicted in Figure 11. K562 cancer cells (same as above) were treated with 1 µM drug concentration for 1 hr at 37 °C and washed twice with buffer by centrifugation. For the SERS measurements, the treated cells were then incubated with pre-aggregated silver colloid for 15 minutes. Non-penetrated aggregates were eliminated by successive washing in the buffer. Before the SERS measurements, the cells were tested for viability (95%). After treatment of a cell population with the drug and incubation with colloids (step A), one cell is selected under the microscope and spectra are recorded at regular intervals along a line (step B). This line of spectra is shown in step C, where one axis represents the frequency domain (cm-1) and the other the points on the line. A different line is then recorded (either by a scanning laser or by moving the XY stage by 1-2 µm intervals). This line will give another pattern as in step C and all the lines are represented in step D, where one square corresponds to a line.
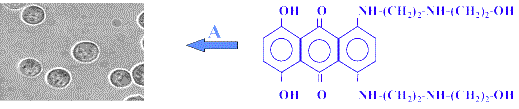
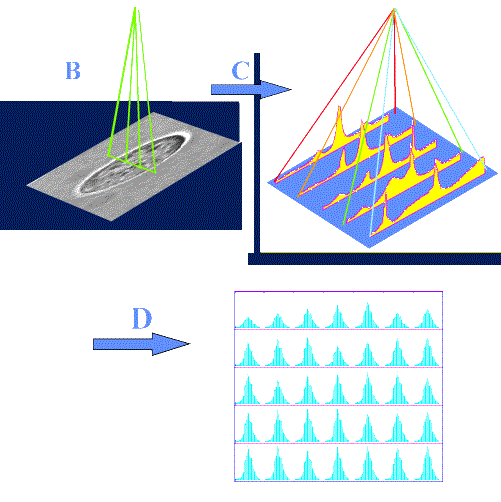
Figure 11. Recording of spectral images from a single K562 live cell treated with MTX
This set can then be used for spectral imaging by exploiting either the whole spectrum range or part of it (functional imaging) utilising either peak intensities, band areas or peak ratios. Figure 12 displays the conventional (left box) and confocal Raman-SERS (centre box) image of a treated K562 live cancer cell. The spectral image is constructed using the area of the band in the 1296-1306 cm-1 region. The white light image clearly shows the presence of silver colloids (dark spots) inside the cell. In the spectral image, the regions of high intensity seem to correspond to the regions where the colloids can be found in the cell. The right box displays two spectra: top – SERS spectrum of free mitoxantrone (5×10-8 M), recorded from a silver colloidal aggregate in the absence of cells; bottom – one of the spectra from the spectral image (nucleus of the cell). The intracellular spectrum exhibits a decrease of the 1308/1270 cm-1 band ratio. The same spectral changes were observed for the model mitoxantrone-DNA complexes studied in vitro [21].
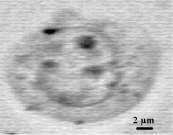
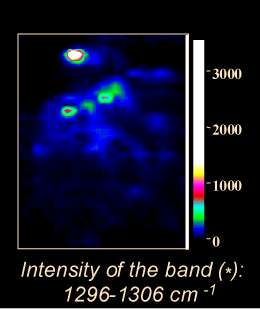
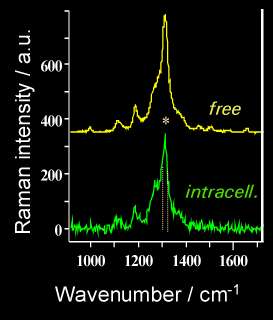
Figure 12. Raman-SERS confocal image of a K562 living cancer cell treated with Mitoxantrone
To summarise: the spectral image has been obtained with confocal SERS microspectroscopy from single, live K562 cancer cells treated with an anticancer drug, mitoxantrone, at a concentration of 10-7M. SERS spectra have been recorded from the inside of the cell in which silver colloidal particles have been introduced to generate the SERS effect. First observation: a satisfactory S/N ratio is obtained although the laser power is incredibly weak (0.1 mW, i.e., 100 times less than in conventional Raman microspectroscopy). This range of power is quite acceptable for live cells, as it does not affect their viability (no hole in the cell!!). Second observation: with respect to classical SERS microspectroscopy, SERS spectral imaging offers better sampling of the spectral information coming from a given localisation. The superposition of the spectral image with a conventional image allows to attribute the spectral information to different cell compartments. The spectral image in this example was constructed following the distribution of the intensity of the mitoxantrone band at 1300 cm-1enhanced by the colloidal particles. The advantage of spectral imaging is that other representations can be used: band integral intensity, band ratios, functional groups etc.., and each time the information can be retrieved easily by clicking on any part of the image. The use of pseudo-colours can facilitate the image representation and in this case it is easier to see how the drug behaves in different cell compartments, e.g., bound to DNA in the nucleus, free in the cytoplasm…The potential of confocal SERS microspectroscopic imaging at the single cell level is very important for the selective analysis of drugs inside these cells.
Some perspectives
The high sensitivity and molecular specificity of SERS microscopy and the confocal property of the instrument give us the possibility of generating multidimensional (space, frequency, time…) spectral images with high spatial resolution. Each dimension of the recorded data corresponds to a specific investigated feature, e.g., localisation of a drug (x, y, z), characterisation (u ), kinetics (s) or temperature (T). However, the SERS approach to intracellular investigation can be improved. So far, we have seen that the colloid particles can be introduced inside the cell by endocytosis or mixing. This introduction is random and can not be totally controlled by the investigator. Another possibility is to use point probes and to deposit, by perforation, the colloidal particles in any cell compartment using a micro-capillary. Cells are not irreversibly damaged by inserting the micro tip. Microelectrodes can also be used as point probes as they can easily perforate the cell and the SERS signal could then be recorded from any given point. SERS spectral imaging is done in the vicinity of the point probe and this enables a statistical analysis of the spectra. SERS spectral imaging could be done on larger areas by using area substrates. Thus using vacuum-deposited metal island films where, in principle, scanning the laser over the whole cell should result into enhanced signals from any point in the cell; in opposition to colloids, which are point probes, and their distribution being random (unless deposited by a microcapillary), enhanced signals are collected only from their vicinity. As we have explained before, island films can be potential candidates for such studies since they act as an external area probes. The combination of gold island films and NIR lasers (dispersive instruments and CCDs) should suit cellular level studies if the chromophore can benefit from long range enhancement. However, as we have mentioned care should be taken to ensure the origin of the SERS signal. Finally, the availability of PMTs which work in the NIR will breath new life into the use of Raman and increase the sensitivity of the technique for cell studies.
References
- M. Manfait, L. Bernard and T. Theophanides, J. Raman Spectrosc., 11, 68 (1981).
- G. Smulevich, A. Feis, A.R. Mantini, M. P. Marzocchi, Indian J. of Pure and Applied Physics, 26, 207 (1988).
- M. Fleischmann, P. J. Hendra, and A. J. Mcquillan, Chem. Phys. Lett., 26, 123 (1974).
- D. J. Jeanmaire and R. P. vanDuyne, J. Electroanal. Chem., 84, 1 (1977).
- R. P. Van Duyne, K. L. Haller and R. I. Altkorn, Chem. Phys. Lett., 126, 190 (1986).
- K. L. Norrod, L. M. Sudnik, D. Roussel, and K. L. Rowlen, Applied Spectrosc., 51, 994 (1997).
- T. M. Cotton, J. H. Kim, and G. D. Chumanov, J. Raman Spectrosc., 22, 729 (1991).
- M. Manfait, H. Morjani, R. Efremov, J. F. Angiboust, M. Polissiou, and I. Nabiev, in The Royal Society of Chemistry, ed. by R E. Hester and R. B. Girling, York, UK, 303-304 (1991).
- Nabiev, H. Morjani, and M. Manfait, Eur. Biophys. J., 19, 311 (1991).
- G. D. Sockalingum, I. Chourpa, J. M. Millot, I. Nabiev, S. Sharanov, and M. Manfait, in Biomedical applications of Spectroscopy, ed. By R. J. H. Clark and R. E. Hester, Wiley, Chichester, 49-87 (1996).
- P. C. Lee and D. Meisel, J. Phys. Chem., 86, 3391 (1982).
- J. A. Creighton, C. G. Blatchford, and M. G. Albrecht, J. Chem. Soc. Faraday II, 75, 790-798 (1979).
- A. Beljebbar, G. D. Sockalingum, H. Morjani, J. F. Angiboust, M. Manfait, Spectrochim. Acta Part A, 53, 123, (1997).
- T. R. Breitman, , S. E. Selonick, , and S. J. Collins, Proc. Natl. Acad. Sci.,USA, 77, 2936 – 2940 (1980).
- T. R. Breitman, , S. J. Collins, and B. R. Keene, Blood, 57, 1000 – 1004 (1981).
- M. Petkovich, N. J. Brand, A. Krust, and P. Chambon,. Nature, 330, 444 (1987).
- C. Favard, P.Valisa, M.Egret-Charlier, S.Sharanov, C. Herben, M. Manfait, E. da Silva, P. Vigny, Biospectroscopy, 5, 101, (1999).
- P. Dhamelincourt, J. Barbillat and M. Delhaye, Spectroscopie Europe, 5/2,16, (1993).
- S. Sharonov, I. Chourpa, P.Valisa, F.Fleury, A. Feofanov, M. .Manfait, Miscroscopy and Analysis, 4, 23, (1994 ).
- I. Chourpa, H. Morjani, J.F. Riou, M. Manfait, FEBS Lett., 397, 61, (1996).
- I. Nabiev, A. Baranov, I. Chourpa, A. Beljebbar, G.D. Sockalingum, M. Manfait, J. Phys. Chem., 99, 1608, (1995).
- I. Chourpa and M. Manfait, J. Raman Spectrosc., 26, 813, (1995).
- I. Nabiev, I. Chourpa, J.F. Riou, C.H. Nguyen, F. Lavelle, M. Manfait, Biochemistry, 33, 9013, (1994).
- J. R. Brown and S. Heider Imam, Prog. Med. Chem., 21, 196, (1984).
- A. Beljebbar, G. D. Sockalingum, J. F. Angiboust, M. Manfait, Appl. Spectrosc., 50, 148, (1996).
REF: G.D. Sockalingum, S.Charonov, A. Beljebbar, H. Morjani, M. Manfait & I. Chourpa Int. J. Vib. Spect., [www.irdg.org/ijvs] 3, 5, 3 (1999)