Non-destructive identification of minerals by Raman microscopy
2. Non-destructive identification of minerals
by Raman microscopy
Ray Frost, Theo Kloprogge, and Jolene Schmidt
Centre for Instrumental and Developmental Chemistry,
Queensland University of Technology,
Brisbane, Queensland,
Australia
Introduction
The identification of minerals is, in many cases, based on techniques like X-ray diffraction, optical microscopy and electron microprobe analysis. A major disadvantage of these techniques is that the mineral crystals have to be destroyed either to a powder or a thin section. Raman spectroscopy has been used for the identification of minerals [1-5]. This paper describes the application of Raman microscopy [6] as a non-destructive technique for the identification of minerals, suitable also for single crystals as small as a few hundred micrometers [7].
The identification of minerals generally starts in the field by visual examination by a geologist. Based on properties such as colour, crystal habitus, hardness, lustre, cleavage, etc. often in combination with detailed knowledge of the regional geological history a first identification of many rock-forming minerals is possible. It becomes more difficult for the more rare minerals mostly present in only minor amounts and often as small crystals. In many of these cases visual examination even by an experienced mineralogist does not give a definite answer and other analytical techniques are needed in order to come to an identification of a mineral. The classical petrologist or mineralogist will start with the preparation of thin sections of approximately 30 microns thickness of the rocks sampled in the field for examination under the optical microscope. Optical properties such as colour, pleochroism, refractive index, birefringes, etc. allows minerals to be identified. However, this method is rather timeconsuming and depends strongly on the experience of the geologist. Alternatively, minerals can be identified by their crystal structure by X-ray diffraction and chemical composition by methods like electron microprobe analysis. In general, X-ray diffraction uses powdered samples, whereas the electron microprobe uses thin sections similar to those for optical microscopy, but without coverglass and coated with carbon. This can pose a problem for the identification of very small or rare crystals. An interesting non-destructive alternative is a spectroscopic technique generally known as Raman microscopy. In this paper we report a number of spectra of various minerals, all with crystals not larger than a few millimetres. This will show the strength of the Raman microscopy technique as it will show that each spectrum is unique and can be used as a sort of fingerprint for the identification of the mineral when a large enough database of mineral spectra is available.
Raman microprobe spectroscopy
The Raman effect is a light scattering effect . In order to obtain the Raman spectra of minerals, minerals were placed on a polished stainless steel surface on the stage of an Olympus BHSM microscope, equipped with 5x, 20x and 50x objective lenses. No sample preparation was needed. The microscope is part of a Renishaw 1000 Raman microscope system, which also includes a monochromator, a filter system and a charge coupled device (CCD). Raman spectra were excited by a Spectra-Physics model 127 HeNe laser (633 nm), recorded at a resolution of 2 cm-1in sections of 1000 cm-1 for 633 nm excitation. Repeated acquisitions using the highest magnification, were accumulated to improve the signal to noise ratio in the spectra. For the 298 K spectra, data were collected at 20-second intervals for 10 minutes at maximum magnification. Spectra were calibrated using the 520.5 cm-1line of a silicon wafer. It should also be noted that the filters in the Renishaw spectrometer start to eliminate the Rayleigh line at about 150 cm-1. This makes the determination of bands below 200 cm-1 difficult and without reliability.
Spectra obtained with the microprobe normally consist of Raman bands superimposed on a background, which is a combination of the fluorescence and the instrument function. In the case of the HeNe laser and the measurement of the hydroxyl stretching region, the background is predominantly due to fluorescence and consists of a sloping linear baseline that is easily corrected. For the HeNe laser and the determination of the low frequency region, the background is predominantly a combination of the instrument function that is dependent on the behaviour of the notch filters, the grating-detector responses, and the fluorescent background. This background is induced by elements placed in the optical path between the sample and the entrance slit of the spectrometer. The background, although a complex function, is easily measured using the incandescent white light from the microscope. The measured spectra are then ratioed to this instrumental background to give the spectra as illustrated in the figures in this paper. Such a method ensures the correct instrumental function is taken into account and that the true spectrum is measured. Different instrumental profiles are obtained for the operation of different lasers.
Normally with conventional dispersive Raman spectroscopy and to lesser extent, Fourier transform Raman spectroscopy data collection times can be excessively long, particularly for minerals. Often for poorly scattering minerals such as clays, several thousand FT Raman spectra must be coadded to obtain a spectrum of sufficient quality. In the case of Raman microprobe spectroscopy with multiplex detection, data collection times are very short and spectra are easily obtained within 1 to 10 minutes. Typically a collection time of 60 seconds, in which 10 spectra are co-added, is used with the maximum magnification. Power at the sample is in the 0.1 to 1 mW range. This use of such low power is a major advantage particularly over that used for FT Raman spectroscopy where typically the power used for minerals is 100 mW for a spot size of 200 m m. For Raman microscopy, the laser spot size is 0.8 microns and the laser power is between 1 and 4mW. Such low power means that there will be no heating effects and consequential damage to the mineral. It was found that the best quality spectra were obtained by scraping fresh crystals from a lump of the raw material on the metal support. Crystals of the kaolinite are easily observed under the microscope.
One of the disadvantages of Raman microprobe analysis is the fluorescence of the clay minerals which is particularly pronounced when using the HeNe laser at 633 nm and other lasers of similar wavelengths. Each of the laser lines used to excite the Raman spectra of the kaolinite polymorphs caused fluorescence that subsided with time. Even so difficulty of measuring the low frequency region with the 633 nm excitation was experienced and the use of the diode laser operating at 780 nm did not alleviate the problem. Obtaining spectra at 77K, meant that the instrumental background shifted with temperature. This shift may be related to the change in the fluorescence of the sample at liquid nitrogen temperatures. Unique spectra from kaolinite polymorph crystals were more difficult to obtain from ground or powdered samples. If the crystals are smaller than the resolution of the microscope then spectra are obtained from an area that covers several crystals and an averaged spectrum is obtained.
An advantage of Raman spectroscopy is that bands in the 200 to 400 cm-1region are easily measured. Dispersive Raman spectroscopy inherently has the clay spectrum superimposed on the intense Rayleigh line. The use of Raman microscopy employing CCD detectors is greatly advantageous because of signal to noise improvement. Detectors in both FT Raman spectrometers and conventional dispersive instruments are photon noise limited. With a CCD detector this is no longer true: typically the noise from a CCD detector is at least 100 times less than from a semiconductor detector. The disadvantage of using Raman spectroscopy operating at the diode laser frequency of 780 nm is the loss of scattering efficiency. Raman scattering decreases as the fourth power of the wavelength of the scattered radiation, so the intensity of the Raman scattering at 780 nm compared to say 532 nm is ~7 times less. For the 633 nm (HeNe) laser, this factor is 2. Nevertheless, the disadvantage of this loss of efficiency is greatly outweighed by the advantages of the CCD detector. Further the advantage of using lasers operating in the 633 or 780 nm range rests with the reduction of the laser induced fluorescence which is a problem when using green or blue excitation wavelengths.
Classification of minerals based on crystal-chemical composition and their Raman characteristics
Minerals can be described as naturally occurring, inorganic crystalline materials. They can be classified based on their chemical characteristics. In general the following eight classes can be identified [8]:
- Elements
- Sulphides and related compounds
- Halides
- Oxides and hydroxides
- Carbonates, nitrates, borates
- Sulfates, tungstates, chromates and molybdates
- Phosphates, arsenates and vanadates
- Silicates
|
Sulphides |
<500 cm-1 |
|||
|
Hydroxides |
3000-4000 cm-1 |
600-1200 cm-1 |
||
|
oxides |
<1200 cm-1 |
|||
|
Carbonates |
1300-1550 cm-1 |
800-890 cm-1 |
670-700 cm-1 |
|
|
Nitrates |
~ 1050 cm-1 |
800-850 cm-1 |
715-770 cm-1 |
|
|
Borates |
1250-1350 cm-1 |
600-900 cm-1 |
||
|
Sulphates |
900-1250 cm-1 |
570-680 cm-1 |
||
|
Tungstates |
750-950 cm-1 |
250-450 cm-1 |
||
|
Chromates |
800-950 cm-1 |
350-500 cm-1 |
||
|
Molybdates |
750-950 cm-1 |
250-450 cm-1 |
||
|
Phosphates |
900-1150 cm-1 |
400-600 cm-1 |
||
|
Arsenates |
770-900 cm-1 |
350-400 cm-1 |
||
|
Vanadates |
700-900 cm-1 |
300-400 cm-1 |
||
|
silicates |
800-1200 cm-1 |
Table 1. Characteristic frequency regions for various anionic groups.
An important part of this classification is based on the occurrence of various characteristic anionic groups. Each of these groups is reflected as specific bands in both the infrared [9] and Raman spectra [10] and function as fingerprints for specific minerals. The characteristic frequency regions of the various characteristic groups are listed in Table 1. It has to be kept in mind that the following description is only very general and that in specific cases the situation can be much more complex. For example distortion of a symmetric anionic group in the crystal structure can result in a splitting of the Raman bands.
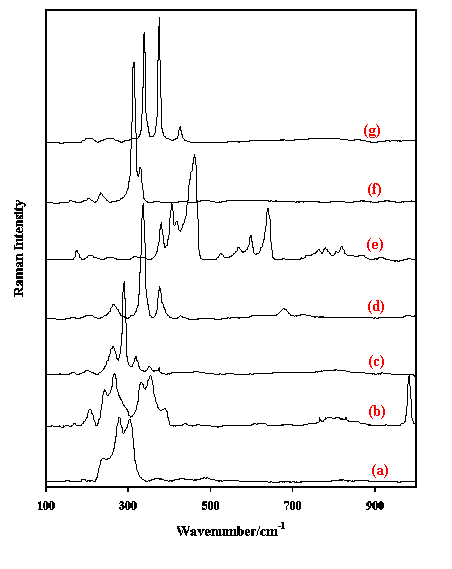
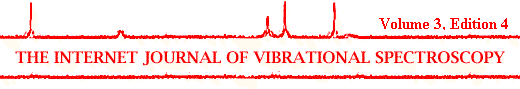
Figure 1. Raman spectra in the region 100-1000 cm-1 of (a) antimonite, (b) cobaltite, (c) chalcopyrite, (d) enargite, (e) molybdenite, (f) kermesite and (g) pyrite.
The sulphides are characterised by the metal-sulphur bonds giving characteristic bands in the low frequency region below 500 cm-1. Figure 1 shows the low frequency region of antimonite (Sb2S3), cobaltite (CoAsS), chalcopyrite (CuFeS2), enargite (Cu3AsS4), molybdenite (MoS2), kermesite (Sb2S2O) and the well-known pyrite or fools gold (FeS2). Although all minerals show the expected bands in the region below 500 cm-1, all minerals still show their own characteristic pattern useful for identification. Hydroxides give rise to hydroxyl stretching bands in the region between 3000 and 4000 cm-1, accompanied by bending modes around 600-1200 cm-1. The presence of water in the crystal structure is reflected in bands around 3200-3500 cm-1 and around 1600-1650 cm-1. The metal-oxygen bonds give rise to bands in mainly the low frequency region below 1200 cm-1. The exact position of the various metal-oxygen vibrations in the spectrum depends on factors like which metal is involved (atomic weight) and the bondlength and bond strength.
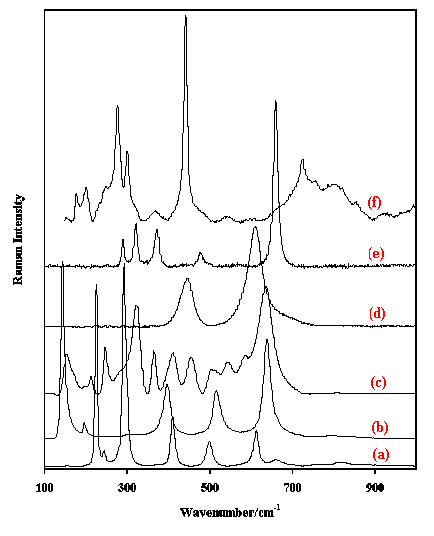
Figure 2. Raman spectra in the region 100-1000 cm-1 of (a) tantalite, (b) anatase, (c) brookite, (d) ilmenite, (e) jacobsite and (f) brucite.
Figure 2 shows a few examples of oxide minerals like tantalite ((Fe2+,Mn)(Ta,Nb)2O6), anatase and brookite (both TiO2), ilmenite (FeTiO3), jacobsite ((Mn2+,Fe2+,Mg)(Fe3+,Mn3+)2O4) and the hydroxide brucite (Mg(OH)2). The carbonates around characterised by three major regions between approximately 1300-1550, 800-890 and 670-760 cm-1.
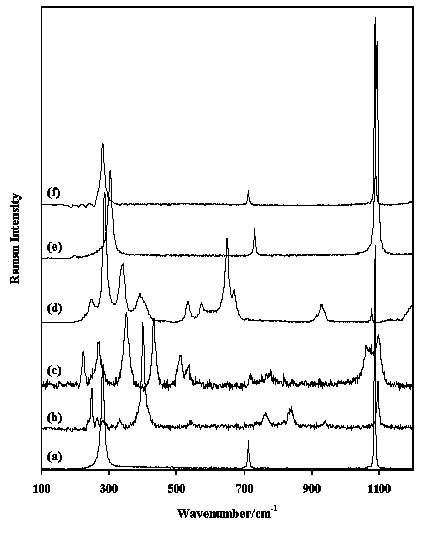
Figure 3. Raman spectra in the region 100-1200 cm-1 of (a) calcite, (b) azurite, (c) malachite, (d) gaudefroyite, (e) smithsonite and (f) strontianite.
In Figure 3 the Raman spectra of some well-known and less well-known carbonate minerals are shown. These minerals include calcite (CaCO3), azurite (Cu3(CO3)2(OH)2), malachite (Cu2(CO3)(OH)2), gaudefroyite (Ca4(Mn3+)3-x(BO3)3(CO3)(OH,O)3), smithsonite (ZnCO3) and strontianite (SrCO3). Borates can be recognised by bands in the regions 1250-1350 cm-1 and 600-900 cm-1. Where borate bands are observed depends on the mineral structure and on the type of borate (metaborate BO22-or orthoborate BO33-).
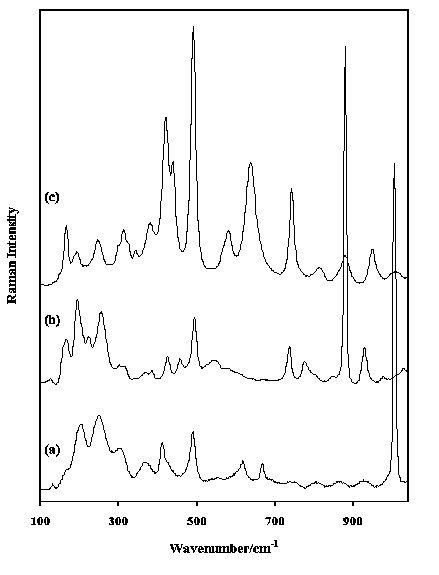
Figure 4. Raman spectra in the region 100-1000 cm-1 of (a) ulexite, (b) kernite and (c) inderite.
Figure 4 shows the low frequency spectra of ulexite (NaCaB5O9.8H2O), kernite (Na2B4O7.4H2O) and inderite (MgB3O3(OH)5.nH2O). Ulexite and kernite show a typical borate band around 1007 and 880 cm-1. In addition both kernite and inderite show two bands around 1350 and 1390 cm-1. Nitrates show strong stretching modes around 1050 cm-1 accompanied by bending modes in the regions between 800-850 and 715-770 cm-1. The sulphates are characterised by strong bands in the region 900-1250 cm-1. Frequently they are accompanied by a doublet in the region
570-680 cm-1. These sulphate regions are easily recognised in the spectra in Figure 5 of gypsum (CaSO4.2H2O), its waterfree equivalent anhydrite (CaSO4), hanksite (KNa22(SO4)9(CO3)2Cl), linarite (PbCu(SO4)(OH)2) and kroehnkite (Na2Cu(SO4)2.2H2O).
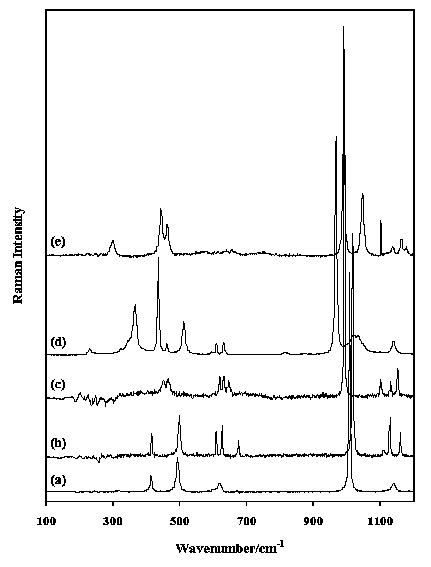
Figure 5. Raman spectra in the region 100-1200 cm-1 of (a) gypsum, (b) anhydrite, (c) hanksite, (d) linarite and (e) kroehnkite.
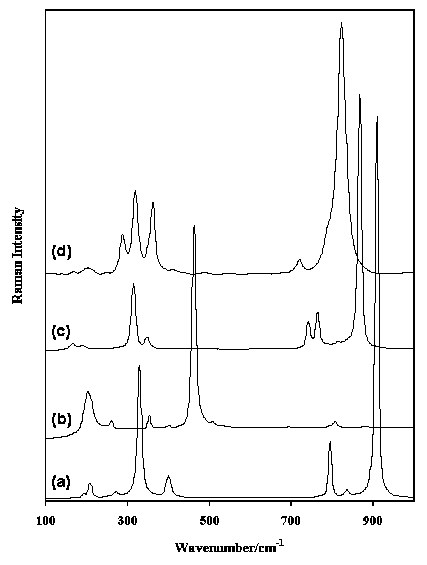
Figure 6. Raman spectra in the region 100-1000 cm-1 of (a) scheelite, (b) wolframite, (c) wulfenite and (d) vanadinite.
Tungstates and molybdates show bands in approximately the same regions around 750-950 cm-1 and 250-450 cm-1, whereas chromates show up in the spectra only at the high frequency sides of these regions (800-950 and 350-500 cm-1). Figure 6 shows the spectra of the tungstates scheelite (CaWO4) and wolframite ([Fe2+,Mn]WO4) and the molybdate wulfenite (PbMoO4). The bands in the regions listed above are easily recognised. Between phosphates and arsenates there are considerable similarities between their physical configurations of the vibrations although they occur in different frequency regions (phosphate 950-1150, 920-970 and
400-600 cm-1, arsenate 770-900 and 350-400 cm-1), while those of arsenate and vanadate are quite different.
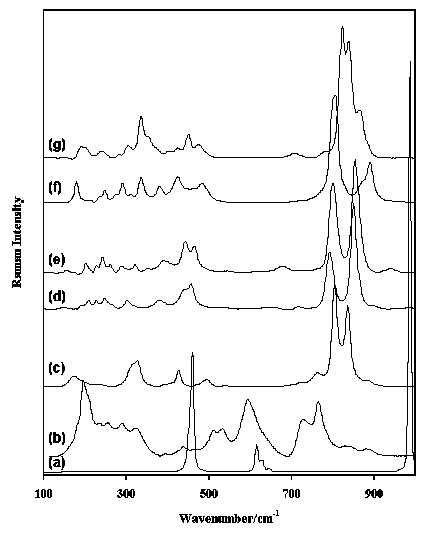
Figure 7. Raman spectra in the region 100-1000 cm-1 of (a) olivinite, (b) cafarsite, (c) bayldonite, (d) erythrine, (e) annabergite, (f) scorodite and (g) roselite.
Figure 7 shows the spectra of some selected arsenates, olivenite (Cu2(AsO4)(OH)), bayldonite (PbCu3(AsO4)2(OH)2), erythrine (Co3(AsO4)2.8H2O), annabergite (Ni3(AsO4)2.8H2O), scorodite (Fe3+AsO4.2H2O)and roselite (Ca2(Co, Mg)(AsO4)2.2H2O), and one arsenite (cafarsite (CaO)8(Ti, Fe)6O10(As2O3)6.2H2O). Strong arsenate bands are easily recognised in the region between 750 and 900 cm-1. This figure also shows the sharp difference between arsenates and arsenites. Vanadates have characteristic bands in the regions 700-900, around 870 and 300-400 cm-1. Figure 6 shows the spectrum of vanadinite (Pb5(VO4)3Cl) with a strong complex band around 800 cm-1 and multiple bands in the region between 275 and 400 cm-1. The silicates form the largest group of minerals on earth. The silicates can be divided in four major groups based on how the SiO4 units are linked together in the crystalstructure: ortho and ring silicates (e.g. olivine group, garnet group, tourmaline, beryl, etc.), chain silicates (e.g. pyroxene and amphibole groups), sheet silicates (e.g. clays and micas) and framework silicates (e.g. quartz, feldspars, zeolites, etc.). All silicates show very strong bands in the region between approximately 800 and 1200 cm-1, although the environment of the SiO4 group affects the shape and position of the various bands.
Conclusions
Raman microscopy has proved an extremely useful technique for the analysis of minerals. The use of the 633nm laser and the X50 objective proved the most useful combination. Fluorescence is not a problem for many minerals, and may be overcome through background subtraction. Impurities of lanthanides and actinides may cause this fluorescence which may prevent the collection of data. One of the difficulties is the collection of low intensity spectra on a large intense background. The collection of data for extended periods of time up to 30 minutes can overcome this difficulty. Nevertheless the advantages of the technique greatly outweigh the disadvantages, and the technique of Raman microscopy to the study of minerals offers a somewhat new and novel technique for the study of mineral chemistry.
It can be said that Raman microscopy is a very powerful tool for the identification of minerals. It offers the possibility to identify minerals based on their specific Raman spectrum, which is unique for every mineral. In addition useful information is gathered about the presence of specific anionic groups and bonds like metal-oxygen or metal-hydroxyl bonds. Raman microscopy is a useful technique for the analysis of even very small crystals still present in handspecimens collected in the field, single crystals or even on uncovered thin sections like the ones used for electron microprobe analysis.
Acknowledgments
The financial and infra-structural support of the Queensland University of Technology, Centre for Instrumental and Developmental Chemistry is gratefully acknowledged.
References
- E. Coleyshaw, W.P. Griffiths and R.J. Bowell, Spectrochomica Acta 50A, (1994) 1909.
- R. L. Frost, J.R. Bartlett and P.M. Fredericks, Spectrochimica Acta 49A, (1993) 667.
- R. L. Frost and S. J. Van der Gaast, Clay Minerals 32 (1997) 471.
- A.G. Smallwood, P.S. Thomas, A.S. Ray, Spectrochimica Acta 53 (1997) 2341.
- J.M. Alia, Y. Diaz de Mera, H.G.M. Edwards, P.G. Martin, S.L. Andres, Spectrochimica Acta 53 (1997) 2347.
- P.J. Hendra, Internet J. Vib. Spec.[www.ijvs.com] 2, 3, 3 (1998)
- R.L. Frost, Chemistry in Australia, 63, (1996) 446.
- J.T. Kloprogge, and R.L. Frost, Klei, Glas en Keramiek, 19 (1998) 22.
- P. Ramdohr and H. Strunz, Klockmann’s Lehrbuch der Mineralogie, 1967.
- J.A Gadsden,., Infrared Spectra of Minerals and Related Compounds, 1975.
- S.D. Ross, Inorganic Infrared and Raman Spectra, 1972.
REF: Frost R., Kloprogge T., & Schmidt, Int. J. Vib. Spect., [www.irdg.org/ijvs] 3, 4, 2 (1999)