Synchrotron infrared microspectroscopy applied to petrography in micron range scale
Guilhaumou*(1), P . Dumas (2), J. Ingrin (4)
G. L. Carr, (3) and G. P. Williams (3)
(1) – C.N.R.S.,URA 1759,
Departement de Géotectonique-Université Pierre & Marie Curie,
4 place Jussieu, 75252 Paris, Cedex 05. France.
(2) – LURE and LASIR-CNRS,
Centre Universitaire Paris-Sud, F 91404-Orsay, Cédex, France
(3) – National Synchrotron Light Source,
Brookhaven National Laboratory, Upton, NY 11973. USA.
(4) -Laboratoire de Minéralogie, UMR 5563,
39 allées Jules Guesde, 31000 Toulouse, France.
Abstract
Thanks to the intrinsicly high brightness of the synchrotron infrared source, areas as small as a few micrometres in dimension can be probed successfully in any kind of minerals providing high contrast direct microanalyses of the in situ fluid entrapped. Several novel applications to petrography were performed to investigate the potentiality of this method for the detection of volatiles and chemical components in these so-called fluid inclusions. We have obtained fingerprints of organic compounds entrapped under geological pressure conditions in reservoir cements, thus allowing us to study petroleum evolution during maturation and migration. High signal to noise ratio spectra allow semiquantitative viewing of fluid inclusions, but refraction effects preclude at the present time precise quantitative analysis of the volume entrapped. Low detection levels of aliphatics, aromatics, CO2 and H2O pinpoint some heterogeneities during fluid migration. CO2 and H2O are easily detected either in the vitreous or gaseous part of glass melt fluid inclusions and are representative of the composition of the magma. Diffusion coefficients for hydrogen is accurately measured from short hydrogen concentration profiles obtained easily in nominally anhydrous minerals.
Progress in petrography over the last few decades has made it clear that most rock formation and subsequent transformation involves the fluid phase. Whether the fluid is a silicate magma, a water solution and/or a mixture of a gas or hydrocarbon, a fluid will be implicated in the chemical processes and in mineral crystallisation and its transformation. It now seems that CO2+H2O are crucial in the mechanism of magmatic fractionation during the evolution of the earth’’s mantel. Hence the challenge exists for contemporary petrologists to analyse minerals at the micron level and to search for evidence of fluids. Using microspectroscopy on unmounted doubly polished thin sections, molecular maps can be made that can be directly correlated with the familiar optical petrographic views. Clearly microspectroscopy is particularly attractive because it is non-destructive and where included fluids are seen, does not disturb the overall structure.
Introduction
Mid infrared is capable of providing highly specific fingerprints of molecular vibrations in the range 600-4000cm-1 in most mineral specimens [1,2]. If the infrared radiation is focussed into samples through an i.r. microscope molecular structural mapping is feasible [3]. However until recently, the classical i.r. microscope and spectrometer allows us to resolve down only to the diffraction limit – in practice to ~20 micrometers [4]. This limitation drastically restricts the application of the method to petrography and particularly so in the study of hydrocarbon inclusions which are frequently of a size less than 10 microns diameter in reservoir cements [5]. Finer resolutions can be obtained if Synchrotron infrared sources are used to illuminate the microscope in a confocal arrangement. To test the application of this technique in the study of petroleum migration, organic species have been analysed inside fluorite matrix and diagenetic quartz removed from petroleum reservoirs. Further, the sensitivity of infrared methods in detecting CO2 + hydroxyl groups (both ÒH and bonded H2O) has enabled us to detect these fragments in naturally quenched glasses within basalts [6] and even to estimate from concentration profiles, diffusion coefficients of OH groups in minerals normally considered to be anhydrous e.g., pyroxene and olivine [7,8]. These preliminary results demonstrate great potential for further developments in the petrography of petroleum bearing occurrences [9].
Instrumentation and Analytical conditions
Instrumentation Experiments were carried out on the U2B beamline at the National Synchrotron Light Source of the Brookhaven National Laboratories in Upton NY, USA. An FTIR infrared micro-spectrometer (Irµs by Spectra Tech) is interfaced to the synchrotron beam [10-13]. The optical system includes an optical module so that the user can identify the zone wherein the spectrum being recorded. An introduction to micro-infrared spectrometry has appeared recently in ijvs [Volume 2, Edition 4].
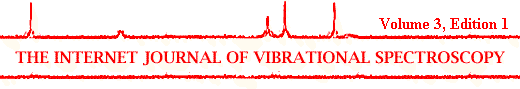
The Synchrotron source produces a beam which when passed through the microscope transmits about 70% of its energy through a pinhole only 12.5 micrometers in diameter (see Figure 1). This contrasts with a conventional blackbody source (often a globar operating at 1500°C) which generates in a similar microscope a patch about 200 micrometers in diameter at the sample stage. The Synchrotron infrared source has a much higher brightness and stability than a black body (typically x102 to 103 in brightness). An account of the properties of the Synchrotron source is detailed earlier in this edition see A.Morris I.J.V.S. 3 1 Section 1.
These advantages are reflected in a considerable increase in the S:N ratio in the recorded spectra. Using a 3 micrometer aperture the advantage is ~x35. The infrared beam is handled using Cassegrain mirror optics X 15 (again see Figure 1) and signal is recorded using a mercury-cadmium-telluride detector.
A colour CCD camera captures a visible image of the specimen enabling us to identify the inclusions under investigation. An automated X-Y mapping stage permits us to scan specimens with a step size accuracy and reproducibility of 1µm. The objective and condenser of the confocal arrangement were both of the Schwarzschild Reflection type. To minimise atmospheric absorption (due to water vapour and CO2) the entire system can be purged throughout using dry nitrogen.
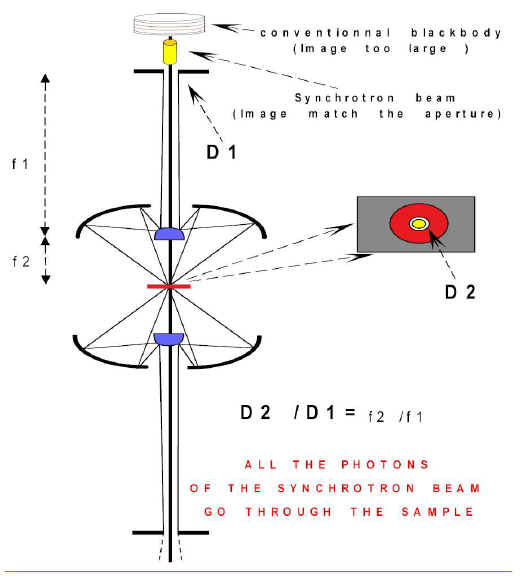 Figure 1. Simplified view of beam interface showing the advantage of the Synchrotron source resulting in high brightness.
Figure 1. Simplified view of beam interface showing the advantage of the Synchrotron source resulting in high brightness.
To study fluid inclusions and quenched glass inclusions doubly unmounted polished slices of the host rock 0.1 to 0.5mm thick were put mounted on a support drilled with a hole 1mm in diameter to avoid using KBr plate . The specimens were mounted on the X-Y microstage. Specimens are analysed in transmission using a 32x (NA 0.65) objective and a 10x (NA 0.71) condenser.
Sampling
It was found that a 3 x 3 micrometre aperture was the most appropriate providing excellent spectra with good S:N ratios. Spectra were recorded at 4cm-1 resolution over 256 co-added scans (requiring a total sampling time of about 110 seconds). Triangular apodization was adopted throughout.
Mapping was achieved using a raster of spectra with a separation of 2 and 3 micrometers. Each spectrum is baseline subtracted to account for the intensity decrease of the of the beam during each photons injection.
Where measurement were made on the OH moieties in the mineral specimens, doubly polished pieces of mineral nearly 1mm thick were mounted over a hole and spectra were recorded as above but with a 10 x 10 micrometre aperture.
Results and discussion
The analysis of the various different inclusions we encounter in minerals has yielded a rich source of information. We have focussed our attention on three main issues.
- Identification of the compounds present in these small volumes whether liquid or gas.
- Variations of concentration of species of interest across interfaces and
- Semi-quantitative analysis of the volume occupied by the various compounds inside an individual inclusion and/or the variation in concentration of one particular chemical compound across the inclusion boundary.
The identification of a fluid
entrapped within a small volume
During dissolution and recrystallisation associated with the production of sediments, fluids are frequently entrapped in small defects in the minerals. These are found as fluid inclusions and are significant because they provide evidence on the paleofluids involved preserving their composition and density through the geological evolutionary process. This is particularly so in cases where transport and re-deposition of ores is involved (e.g., Au, Pb and Zn) and also in petroleum formation [4,5] and migration. The material in filling petroleum reservoirs, the in situ cracking that may have occurred, biodegradation and modification over long periods of oil originally deposited can all be addressed and linked to clay diagenesis [14].
Typical hydrocarbon bearing fluid inclusion inside mineral. Noticed at room temperature the fluid is demixed in liquid and vapor (undercritical state). Liquid water may be present as an immiscible phase but not visible.
Scale = __________ 20µm
Hydrocarbon fluid inclusions in sedimentary settings have been successfully analysed using FTIR microspectroscopy in fluorite and quartz cements and has been related to petroleum migration [15,17]. In silicaceous diagenetic overgrowths, these inclusions are generally less than 10 micrometres in diameter [5]. The oil content of such inclusions is hard to identify but UV fluorescence microscopy is sometimes valuable. On the other hand detecting the presence of water is very difficult as it frequently wets the cavity walls and is simply not visualised by optical microscopy [15,16].The presence of volatiles is important in providing evidence on the formation of organic matter. Components such as dissolved CO2, N2 and SO2 are significant but their presence in an oil inclusion cannot be detected optically.
For the larger inclusions where an infrared fingerprint for oil can be recorded the analysis may be incomplete e.g. aromatic C-H stretching modes have low extinction coefficients hence the detection and quantitation of the aromatic content and distribution are problematic. Unfortunately the location and identification of such species in inclusions are of prime importance for the definition of the overall fluid system [4].
We show below that we have now succeeded in locating and identifying such compounds in fluid inclusions smaller than 20 micrometres.
The occurrence of both petroleum and aqueous fluid inclusions (FI) in siliceous overgrowths from North Sea reservoir sandstone (from the Dunbar area) have been studied previously [18,19]. Two generation of aqueous and organic inclusions were described. The first one (GI) is at the limit between detrital grains and siliceous overgrowths whilst the second one (GII) lie within detrital grains [18]. In GI most of the inclusions are small and less than 10 micrometres in size. In GII the inclusions are sized between 10 and 20 micrometres and are brown in colour. [19] We have performed synchrotron microspectrometric examinations on various inclusions of both types above. We have obtained very precise differentiations within class GI between organic and aqueous fluid containing inclusions and have done so with very high S:N ratios.
In all cases the atmosphere has been used as a background because of the possible heterogeneity of the quartz matrix (presence of inframicroscopic fluid inclusions). The low frequency cut off near 2000cm-1 and the bands at 2133 and 2237cm-1 are due to the quartz itself. The weak and broad features near 2332 and 2372cm-1 are due to atmospheric CO2 and arise from variations in the efficiency of purging.
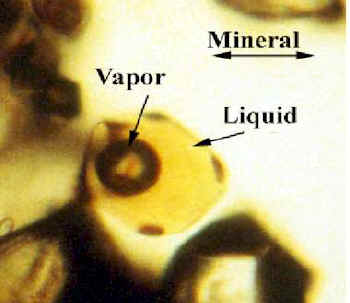
Two types of infrared spectra on inclusions can be recorded. The first type is illustrated in Figure 2 a to d , the second in Figure 3 a to c.

Figure 2. Spectra from four different inclusions within the same context and also the spectrum of quartz under similar spectroscopic conditions. Spectrum 2e is that of quartz matrix (SIO2) in same conditions for comparison (Figure. 2 e). The atmosphere is always taken as background because of the heterogeneity of the quartz matrix in this kind of sample [5]. The low frequency cut off, around 2000 cm-1 and the bands located at 2133 cm-1 and 2237 cm-1 are due to the intrinsic absorption of the quartz. The weak and broad features at 2332 and 2372 cm-1 are due to atmospheric CO2 gas, arising from the variation in the purge content during measurements.
The relative intensity of the CH3 and CH2 characteristics can be used to estimate very approximately the chain length of the aliphatic hydrocarbons. These are estimated to lie in the range C6H14 and C10H22 [16]. A small feature near 3030cm-1 can sometimes be seen although it is very weak. This band might originate from CH4 or aromatics. Methane has been seen in FI inclusions similar to ours using Raman methods [18,19]. The appropriate peak is normally expected to occur near 3010cm-1. Although it is located at higher frequencies 3030cm-1 in the Raman measurements. We therefore propose as a result that the band we observe is of aromatic origin. Carbon dioxide occurs both in the inclusions and in the atmosphere and as a result incomplete purging of the optical path causes confusion. Making appropriate corrections we estimate that the partial pressure of CO2 in the inclusions is around 1 atmosphere.The first type shows a main band centred around 3400cm-1 due to molecular water entrapped inside the inclusions [20-21]. The amount of water varies from inclusion to inclusion. A weak but sharp feature also occurs near 3382cm-1. Its sharpness cannot be associated with molecular water but it can be due to OH groups bonded within quartz [22]. Other inclusions exhibit a broad intense absorption between 2800 and 3100cm-1. The bands seen in Figures 2 and 3 near 2960 and 2875cm-1are due to the asymmetric and symmetric CH stretching modes in CH3 fragments while those at 2925 and 2850cm-1 are due to related motions in CH2 groups [20-21].
Figure 3. shows the spectra of three fluid inclusions of second set (a, b, c) and the spectra in the quartz matrix for comparison (3d), same conditions as in Figure 2.
In the second generation of specimens GII we note two types of inclusion:
- Aliphatic hydrocarbon inclusions containing significant amounts of dissolved CO2 but no H2O [See Figure 3a] and
- a type of inclusion containing both water and oil plus CO2
[Figures 3b and 3c].
The CO2 absorbs at 2338cm-1 suggesting it is associated with oil as a dissolved component. We estimate the aliphatic hydrocarbons have chain lengths between C8and C12 and are free of significant aromatic fraction.
Two formations have been sampled in a sandstone reservoir which presumably have a different geological history from those considered above. In these cases organic fluid inclusions are yellow-brown and are included in quartz overgrowths. Ten out of twenty inclusions in each sample were analysed in the two formations. The characteristic CH stretching bands of aliphatic hydrocarbons and a very strong absorption due to molecular water are seen in Figure 4. The sharp feature near 2337cm-1 in Figure 4b is due to CO2 inside fluid inclusion.
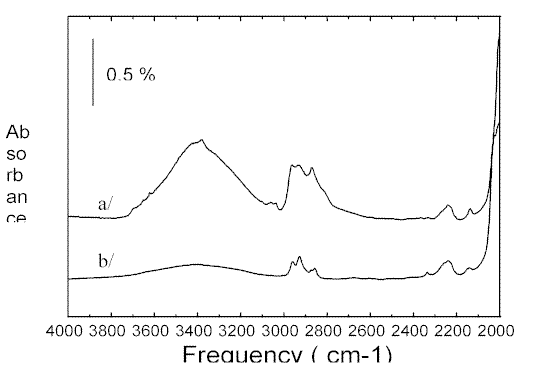
Figure 4. SR-IR spectra of fluid inclusions in a sandstone understhrusted reservoirs a/ and b/ corresponding to two different degree of oil maturation, as revealed by the different vibrational signatures. The spectra result from 400 accumulations, at 4 cm-1resolution, with an aperture of
3×3 µm2
We suggest that the two very different inclusions must be associated with two differing stages in oil maturation.We estimate from the CH2 and CH3 bands in Figure 4b a chain length between C7and C9. Methane is not detected. In Figure 4a a marked feature above 3040cm-1appears due to aromatics but CO2 is not detected. The absorption band pattern in Figure 4a around 2900cm-1 suggests to us that the hydrocarbons are of low molecular weight. We also note narrow peaks at 3620 and 3690 characteristic of hydroxyl groups. Now, they do not appear in Figure 4b so perhaps the OH groups are associated with the aromatic components or of course the aliphatic ones.
Quantitative analysis
Semiquantitaive analyses can be performed exhibiting the change in the integrated intensities of each vibrator inside the fluid inclusions. We have performed such analyses on two different inclusions in fluorite and in quartz in which at room temperature the fluid is undercritical i.e. both a liquid and a vapour phase are present. We first focused on the CO2, water, aliphatic and aromatic vibrational bands. In Figure. 5, we report infrared spectra of typical hydrocarbon entrapped fluid inclusions
A and B, taken with a small aperture (3×3 micrometres). The presence of CO2under pressure, with its characteristic peak at 2338 cm-1 is clearly in evidence in A, as well as the presence of aliphatic components. Hydroxyl groups are also seen at 3592 and 3697 cm-1. We then scanned the inclusion, in a raster fashion (using 3 micrometre steps), in order to record the chemical mapping of the inclusion. We report the image of the chemical composition of the aliphatic compounds, CO2 and the hydroxyl groups (at 3697 cm-1). It is clearly seen that the CO2 is positioned mainly in the vapour phase whilst aliphatics are mainly confined to the liquid. However, an interesting observation is the chemical image of the hydroxyl which match exactly that of the aliphatic compounds. This encourages us in our initial hypothesis that the OH groups form the end groups terminating in the aliphatic compounds, and should motivate further studies to understand the precise chemical nature of these compounds entrapped inside inclusions. In inclusion B typical spectra show aliphatic and aromatic species (band centred at 3030 cm-1) again allowing us to scan the chemical map of their distribution inside mineral.
The quantitative aspect of our analysis of chemical compounds inside fluid inclusions is relevant if one needs to address questions regarding the volume occupied by each chemical compound within an inclusion. However, this type of study is quite difficult. This issue is important in geological analysis, and we want to detail this particular aspect.
One of the properties of IR spectroscopy, is to drawn a linear relationship between the intensity of an absorption band (expressed in absorbance units) and the product of concentration and thickness, for a given vibrator [20-21]. In infrared microspectrometry at and under the diffraction limit one attempts to match the optical image (determined by the size of the projected aperture) and the chemical image. The IR wavelength is of the same order or less than the size of the aperture, hence diffraction effects tend to broaden significantly the projected IR image at the sample stage (Airy diffraction pattern). To reduce this effect and keep a constant lateral resolution for all wavelength recorded, a confocal objective is used (the confocal objective is referred to below as the ‘condenser’). The objective and condenser must be optically conjugated. When a medium is present above and below the inclusion, refraction has to be taken into account in order to keep the objective and condenser conjugated. When achieved, this remains true unless the local shape of the inclusions change, the thickness of the upper and/or lower medium vary, and/or the refractive index of the medium through which the infrared beam propagates does not vary with wavelength. In such a case, the infrared lateral resolution departs from the optical resolution. None of these parameters can be kept under control from one sample to the other, hence errors in quantitation are inevitable. As a consequence, particular caution has to be taken when attempting to extract volume measurements from confocal microscopy in the infrared region.
| footnote: In optics, the diffraction limit d is written: sin(Q) = 1.22.l, where Q is the incidence angle defined by the focusing objective and l the wavelength. The numerical aperture of an objective (N.A.) is n. sin(Q) ( n = 1 for air). So, we end up with d = l / N.A. In the case of a confocal microscope, the diffraction limits becomes d = 1.22 x l / ( N.A. Objective + N.A. Condenser).So typically, d = l. |
Volatiles characterization in magmatic process
CO2 and H2O are particularly important features when deducing the immiscibility characteristics within the mantle because of their implication in the chemical evolution of the magma. These volatiles can be preserved in quenched glass inclusions contained in primary basaltic minerals such as olivine and pyroxenes uplifted as xenocryst during volcanic eruptions. [23]. Their characterisation and location are of widespread interest to those concerned with mineral transport properties and melt formation processes. Typical glass inclusions ( Figure 6 ) are present in olivine matrices e.g., in tholeitic basalts from magmatic eruptions dredged from submerged volcanoes in South Vietnam. They have been previously studied and analyzed by Raman microspectrometry [24]. We have carried out an analysis of some of the inclusions contained in these materials – see Figure 6 where we show a typical spectrum. Interestingly, for this type of inclusion, CO2 (band at 2338 cm-1) and molecular water and OH molecular water, with bands at 3340 and 3510 cm-1 are detected. Further experiments are needed to distinguish between the two species. This analysis is important if we are to understand the origin of the mineral, as the data shown in Figure 5 pinpoints the presence of water in the magma at the time of mineral differentiation and constrains the temperatures involved in the thermal mantle anomaly [23].
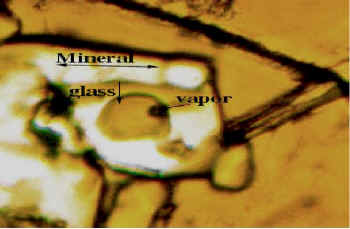
Figure 6. A typical glass melt inclusion inside an olivine matrix and the corresponding infrared spectrum in tholeitic basalts from a magmatic eruption in South Vietnam. The two OH bands at 3340 and 3510 cm-1 correspond to OH and molecular H2O. The stretching band of CO2 is detected at 2338 cm-1.
Scale = __________ 20µm
Hydrogen profile in anhydrous minerals
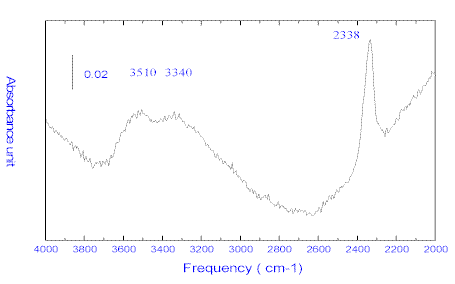
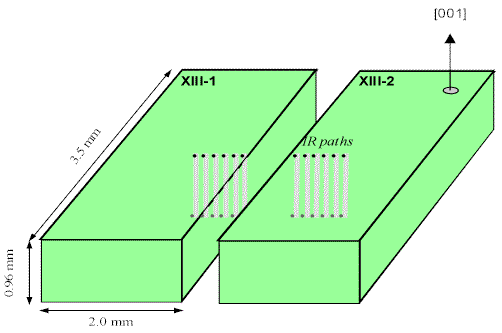
Figure 7. Geometry of the experiment.
The diffusion rate of point defects associated with hydrogen storage in anhydrous minerals like pyroxenes, olivine or garnet (mainly metal vacancies in octahedral sites: VMe) is thought to play a prominent part in the kinetics of hydrogen exchange within the Earth’s upper mantle [25,26]. Among these minerals, diopside is regarded as the most hydrogen-rich within the mantle beneath 410 Km depth (minerals are uplifted during volcanic eruptions) [8]Bell and Rossman, 1992; [27]. Due to the slow self-diffusion of major elements in diopside compared with olivine[28, 29], the diffusion coefficient of the associated point defects in diopside are expected to be significantly lower than those in olivine (coefficient DVMeolivine 10-11 m2s-1) [26].
In order to measure the mobility of the intrinsic point defects involved in the uptake of hydrogen at 1200°C, we have performed the following experiment on a gem quality Russian diopside (Figure 7):
- the single crystal of gem quality diopside was cut into two 0.96 mm thick polished pieces
- one piece (labeled XIII-1) was pre-annealed in air at 1200°C for 72 hours. The two pieces were then heated to 1000°C in 1 atmosphere H2 until saturation of hydrogen was achieved (64 hours at 1000°C).
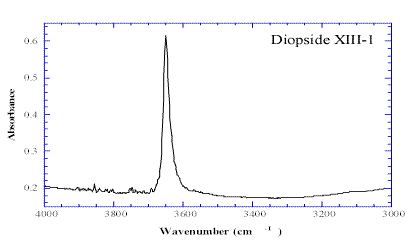
Figure 8. The hydroxyl band observed in the 3000-4000 cm-1frequency region, in the pre-annealed sample of a diopside sample.
Our measured value of the diffusion coefficient at 1200°C, 5±2 x10-13 m2s-1 is more than 20 times smaller than the diffusion coefficient of VMe in San Carlos olivine crystals [26]. It suggests that the diopside equilibration with water within the upper mantle is slower than it is in olivine.Figure 8, shows a typical infrared spectrum, collected on the pre-annealed sample in the 3000-4000 cm-1 frequency region. The hydroxyl peak, at 3648 cm-1, is clearly seen, and its intensity variation has been accurately followed, when stepping from the edge of the two crystals towards the centre (Figure 9). The pre-annealed piece has a hydrogen concentration more than twice that of the hydrogen concentration within the reference piece (labeled XIII-2) and shows a really significant concentration profile. The profile decreasing from the edge toward the centre of the crystal has been estimated and curve fitted assuming an isotropic diffusion in three directions within the sample (Figure 9 see equations in Ref. 30.).
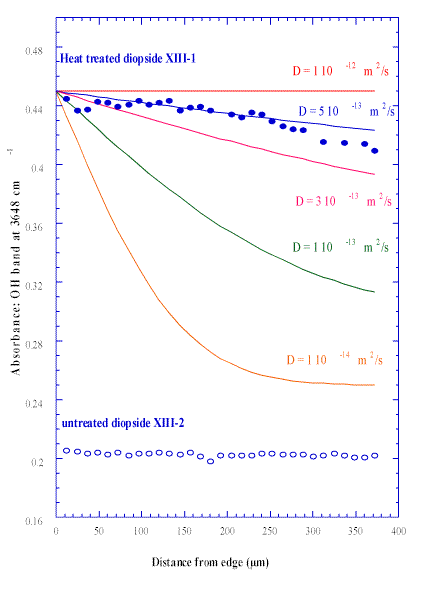
Figure 9. Concentration profiles of hydrogen (the integral of the 3648 cm–1 OH band) away from the samples edge : in a diopside: sample (XIII-I) annealed at 1200ºC, compared with XIII-2: an unannealed sample.
These first results acquired, using synchrotron infrared microspectrometry on a range of samples of geological interest clearly demonstrate that detailed chemical analysis inside small (micron size) inclusions are now feasible, with a high infrared contrast, and therefore open up a range of new investigation domains in geology. For instance, the characterisation of diagenetic processes, particularly useful for reconstitution of palaeofluid evolution in the history of petroleum reservoirs is now possible. A typical example of the application of our methods concern North Sea samples from Dunbar region sandstones. Two oil generations can be differentiated between detrital grains and those derived from in overgrowth zones, suggesting that one of them was formed by refilling of old cavities inside detrital grains and the other one at the beginning of siliceous overgrowth formation [9,19]. Additionally, the heterogeneity of the fluid responsible for the main episode silicification has been clearly evidenced from successive FTIR spectra. In a second example, where a tectonically emplaced reservoir is implicated, the presence of aromatics and dissolved CO2 is identified and considered. Two different oil imprint are identified vis. the differentiation of the maturation level of the oil entrapped during it’’s tectonic history.
Conclusions and perspectives
The CO2 in the vapour phase as well as that dissolved in organic liquids or in the vitreous part of a glass melt fluid inclusion is detected down to a spatial resolution of 3×3 µm2 in low density fluids (< 2 mole %) thanks to the low detection limits and the almost complete purge of the system. The detection of liquid or pressurised vapour water, aliphatic and aromatic components within an inclusion is precise and although as we have explained, of limited quantitative reliability and value, does supply invaluable data on domains less than 10 micrometres in diameter.
Semi quantitative viewing aims to better understand re-partition of the chemical species between liquid and vapor (undercritical) in the cavities and to specify possible oxygenated bonds. The H2O content in silicates may well be observed in traces. Therefore, short OH profiles can easily be measured in anhydrous minerals, providing a means to calculate diffusion coefficient of species involved in the uptake of hydrogen in these minerals.
Acknowledgment
We thank Nathalie Zanier for fruitful discussions on FTIR microspectrometry. We fully acknowledge the technical and engineering support of the NSLS and in particular the help of D. Carlson, J. Gallagher, R. Greene, G. Nintzel, and D. Lynch. We are deeply indebted to John Reffner of Spectra-Tech Inc. for discussions and the NSLS which is supported by the United States Department of Energy under contract DE-AC02-98CH10886.
References
- G.Calas, A.Y.Huc, and B.Pajotj, Bull. Soc. Fr. Minéral. Cristallogr. 99, 153 (1976).
- O.Barres, A.Burneau, J.Dubessy, and M.Pagel, Appl. Spectrosc. 41, N°6, 1000 (1987).
- J. Pironon, J. Sawatzki, and J. Dubessy., Geochim. Cosmochim. Acta, 55, 3885 (1991).
- E.Roedder, Mineral. Soc. Am. Blacksburg 12, 644 (1984).
- R.H.Goldsteins, and T.J.Reynolds, In ” Systematic of fluid inclusions in Diagenetic minerals” SEPM Ed. 199, (1994).
- M.L.Freezotti, Eur. J. Mineral. 6, 805 (1994).
- J. Ingrin, K.Latrous, J.C.Doukhan, and N.Doukan, Eur. J. Mineral. 1, 327 (1989).
- D.R.Bell & G.R.Rossman, Science, 250, 1391 (1992).
- N.Guilhaumou, P.Dumas, G.L.Carr, G.P.William, Applied Spectroscopy,52/8, 1029 (1998).
- G.P.William, Int. J. of Infrared and Millimeter waves 5, 529 (1984).
- G.P.William, Review of Scientific Instrument 63, 1535 (1992).
- G.L.Carr, S.Sutton, R.J.Hemley, and G.P.William, Synchrotron Radiation A 7, 30 (1994).
- L.Carr, J.A.Reffner, and G. P.Williams, Rv. Sci. Instrum. 66, N°2, 1490 (1995).
- J.L.Bantignies, C.Cartier, H.Dexpert, A.M.Flank, and G.P.William: C R Acad Sci Ser II, 320, 8 Part 2 p.699-706 (1995)
- N.Guilhaumou, and N.Szydlowski, C. R. Acad. Sci. 309, Série II, 1171 (1988).
- N.Guilhaumou, N.Szydlowski and B.Pradier, Min. Mag. 54, 311 (1990).
- A.H.Rankin, B.L.Hodge, and M.Moser, Min. Mag. 54, 335 (1990).
- S.Cordon, and N.Guilhaumou, C. R. Acad. Sci. 320, 563 (1995).
- N.Guilhaumou, S.Cordons, C.Durand, and F.Sommer, Eur. J. Miner. (1998), 10, 355.
- G.Herzberg, In « Molecular Spectra and Molecular Structures » D. van Nostrand , Princeton, New Jersey, 13th ed., 308. (1945).
- N.B.Colthup, L.H.Daly, and S.E.Wiberly Academic Press, 548 p. (1990).
- M.Paterson, Bull. Miner. 105, 20 (1982).
- G.Gurenko, and M.Chaussidon, Geochim. Cosmochim. Acta 59, 2905 (1995).
- N.Guilhaumou, and J.C.Touret [1998], to be published
- Q. Bai, & D.L Kohlstedt,. Phys. Chem. Minerals, 19, 460-471 (1993).
- D.L.Kohlstedt, & S.J.Mackwell, Z. Phys. Chemie., 207, 147-162 (1998).
- H. Skogby, Amer. Mineral., 79, 240-249 (1994).
- J.B. Brady and R.H McCallister,. Amer. Mineral., 68, 95-105 (1983).
- A.Dimanov and J.Ingrin Phys. Chem. Minerals, 22, 437-442 (1995).
- J. Ingrin, S. Hercule and T.Charton, J.Geophys.Res. 100, 15489 (1995).
Received in original format 15th January 1999, received in revised format 24th February 1999, accepted 25th February 1999