NIR & Chemometrics: An Application in the Wool Industry
J. S. Church and J. A. O’Neill
Commonwealth Scientific
Industrial Research Organization (CSIRO)
Wool Technology,
PO Box 21, Belmont,
Victoria 3216 Australia
Abstract
Near infrared and advanced data processing such as principle component analysis is being used more and more in industrial environments. In this paper we present a brief introduction to these two techniques through the use of a novel application that we recently developed for the textile industry. The application involves the detection of high density polyethylene and polypropylene contamination in wool. The problem starts out on the farm if small pieces of polymeric material from a wool pack, fertilizer bag, or hay bale twine are picked up with a fleece. If not identified and removed this material shows up as light colored specks in the final fabric. As these hydrocarbon based polymers do not take up wool dyes the effect is particularly pronounced with dark colored fabrics.
Key Words
NIRS, Principal Component Analysis, Polymeric Contamination, Wool
Introduction
Near infrared (NIR) spectroscopy coupled with chemometric methods such as principal components analysis (PCA), principal components regression and partial least squares has seen a rapid increase in use in industrial settings. NIR spectroscopy has been used for both at-line and on-line applications in the textile industry [1-3] including the measurement of residual wool wax, moisture, synthetic / natural fibre blend ratios and the treatment levels for various chemical processes. On-line applications of NIR spectroscopy have been developed in a number of other areas including food quality assurance[4,5], polymer product quality[6], polyol production[7], protein content of yeast cells[8], plastic identification in household waste[9] and quantitative moisture measurements[10].
The contamination of natural fibres with polymeric materials such as high density polyethylene (HDPE) and polypropylene (PP) which originate from wool pack or hay bale twine is a major problem in the wool industry[11]. As these polymers do not take up wool dyes they appear as light colored specks in the final fabric. Recently we have reported on the use of the near infrared spectral region coupled with chemometric methods for the detection of HDPE and PP fibrils entwined in wool and cotton yarns[12]. It would be more efficient however if the polymeric contamination could be removed at an earlier stage of processing. The first step in the processing of wool is that of scouring which removes wool grease, swint and dirt from the wool. It is after this stage that polymeric contaminants are manually removed. This is a tedious process, the success of which depends on human alertness as well as the visibility of the contaminant in the scoured wool. The use of a high speed detector system at the end of the scouring line would decrease the chances of polymeric contaminants continuing through to later stage of processing[13].
In this paper we describe this application to present the basic concepts of NIR spectroscopy and the chemometric method of Principle Components Analysis.
Experimental
Spectroscopy
Spectra were obtained over the range 400 to 1700 nm using a Perten DA-7000 NIR/VIS dual diode array instrument. The sample area analysed is 100mm in diameter and the typical analysis time is 3 seconds. In continuous data collection mode spectrum files can be generated at a rate of the order of one every 1.5 seconds. Data collection was done using Perten DA7000 software (Version 2.00) and data analysis was carried out using Grams/32 (Version 4.04).
Materials
Samples of loose scoured wool, which contained small amounts of vegetable matter and residual wool wax typical of the industry, were obtained from several Australian wool sources. Actual samples of wool bale tape (HDPE), fertilizer bag (PP) and hay bale twine (HDPE) were used as the polymer contaminants.
Methodology
Samples for the static experiments were prepared by spreading 5g of loose wool in a 100mm glass evaporation dish the bottom of which was lined with aluminum foil. This produced a layer of wool roughly 20mm thick, which is typical of that found on conveyors in mills. Samples of polymeric material of varying type, sizes and color were buried at various depths within the wool. Some samples were completely buried so that they were invisible to the eye while others were covered to varying extents.
For the dynamic experiments the loose wool was spread out over a 2m long by 0.2m wide aluminum sheet. The height of the wool pile and its packing density were allowed to vary randomly. Pieces of polymeric material were dispersed along the sheet in a manner similar to that used for the static experiments. The sheet was then pulled past the downward directed NIR beam, at varying speeds with the aid of a motorized winch. This experimental setup, which is shown in Figure 1, mimics the situation at the exit of a production dryer where the wool is transported on a conveyor formed from stainless steel slats.

Figure 1. Photograph of the experimental setup used for the dynamic experiments. The motorized winch is not shown.
Results and Discussion
NIR Spectroscopy
The NIR spectral region has been defined by Kaye [14,15] to extend from 700 to 3500 nm (14285 to 2860 cm-1). The only fundamental absorption bands, those associated with C-H, O-H and N-H stretching, are located between 2500 and 3500 nm. Bands observed below 2500 nm are overtones and combination bands from higher wavelengths[16]. In our recent work[12] we have examined the NIR spectra of hydrocarbon based polymeric materials in wool and cotton yarns between 2250 and 2400 nm. The intensities of vibrations active in this region are relatively strong as they are either first order overtones or combination bands. The diode array spectrometer used in this current study however operates in the 700 to 1700 nm range. This is the spectral region where third and fourth order overtones are active and thus band intensities, especially for thin samples, are much weaker. Working in this wavelength range one can take advantage of the fact that much thicker samples can be analyzed.
The spectra obtained from pure samples of HDPE and loose scoured wool are shown as Figure 2A and B, respectively. From these results it is apparent that the polymeric material and wool are spectroscopically different. The spectrum obtained from a 1mm thick 50mm by 5mm piece of HDPE wool pack buried under a 20mm thick layer of loose scoured wool is shown as Figure 2C. Comparing this spectrum to that obtained from the wool (Figure 2B) it is clear that the detection of polymer materials in the wool in this spectral region is not straightforward. This situation presents an ideal application for chemometric methods, such as principal component analysis [17,18]. Our investigation has been carried out in two phases. Initial work involved the use of spectra collected while the wool and contaminants were static, then in order to more closely simulate the conditions of a wool scouring plant, spectra were recorded continuously while the wool and contaminants were moving under the beam of the spectrometer. These latter experiments also provide some useful insights into how PCA actually works.
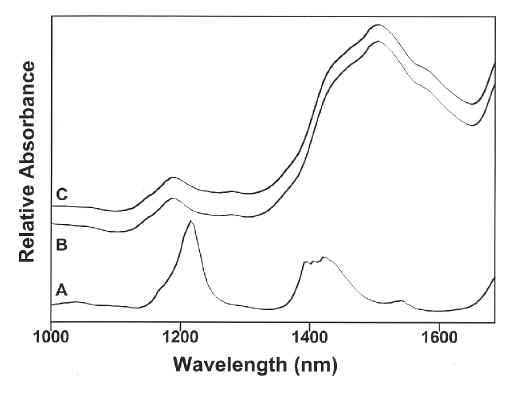
Figure 2. The 1000 to 1700 nm region of the NIR spectra of A) HDPE, B) loose scoured wool and C) a 50mm by 5mm piece of HDPE buried under 20mm of loose scoured wool.
Principal Components Analysis
Principal component analysis is a statistical data analytical technique which has recently seen application in the textile [12, 19-22] and food industries[23]. PCA involves identifying the components accounting for variance within a data set. Averaging the spectral set then comparing this to each spectrum and creating a synthetic spectrum which accounts for the largest part of the variance within the spectral set produces the first loading vector or principal component (PC). The scaling factor that represents the amount of the loading vector in each of the spectra in the data set is known as the score. Multiplying the loading vector by the score for each spectra and subtracting this from the original spectra produces a new spectral set. This process is repeated with the new spectral set until all of the variance is accounted for.
As this type of spectral decomposition uses no chemical information it is not uncommon that the loadings bare little or no resemblance to the pure components present in a multi component sample from which the data was obtained. PCA can be used to determine similarities between various spectra from within the data set. If the score for one PC is plotted against that of another for each member of the data set, separate clusters or groupings may well be produced from spectra such as those obtained from scoured wool and scoured wool with polymeric contaminants.
Static Experiments
Twenty spectra each were collected from scoured wool and the polymeric contaminated wool. The polymeric contaminants were distributed throughout the wool in varying places, as described above, during the collection of spectra. The PCA of the raw spectra was not capable of distinguishing between the wool and the polymeric contaminated wool. It was determined that this was largely due to baseline shifts arising from differences in the way in which the samples are presented for analysis. This is a well known problem which is traditionally overcome through data pre-processing such as first derivative or automatic baseline correction. For our data, the best results were obtained using first derivative spectra from the 1000 to 1700 nm region. The 1st derivative spectra obtained from wool, PP and HDPE are shown as Figure 3A, B and C, respectively.
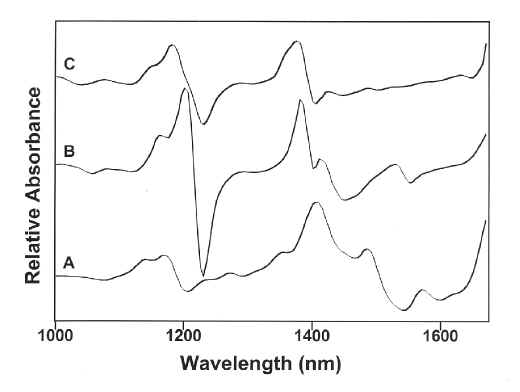
Figure 3. The 1000 to 1700 nm region of the1st derivative NIR spectra of A) wool, B) PP and C) HDPE.
For this model, 4 Principal Components (PCs) account for 95.6% of the total variance (PC1=86.8%, PC2=4.8%, PC3=3.0%, PC4=1.0%). A PC1 versus PC3 scores-scores plot for this data, presented as Figure 4, exhibits two distinct clusters, one of the wool spectra and another containing the spectra of the polymer contaminated wool.
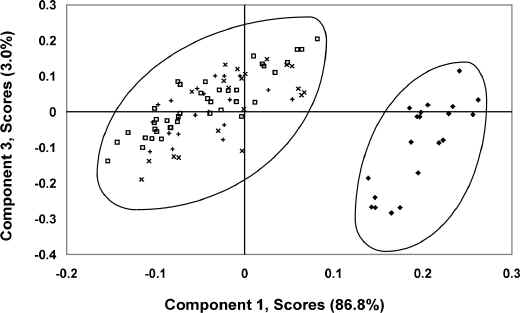
Figure 4. A PC1 versus PC3 scores-scores plot for the 1stderivative, 1000 to 1700 nm region depicting clustering observed for pure and polymeric contaminated wool samples: ( ◆ ) pure wool, ( □) fertilizer bag and wool, ( X ) wool pack and wool and ( + ) hay bale twine and wool. All contaminants were 50mm by 5mm pieces.
The loadings plots, when compared to the spectra obtained from the pure components, can be used to help understand the basis of the separations observed in the scores against scores plots. All of the features observed in the PC1 loadings plot, shown as Figure 5, correspond well with those of the first derivative spectra of pure wool (Figure 3A). The PC3 loadings plot, Figure 6, exhibits a number of intense features that correspond with bands observed in the spectra obtained from the pure components. Based on the work of Wang et al [24], the negative wool feature at 1545 nm can tentatively be assigned to a tertiary combination involving the Amide A mode and the first overtone of the Amide II mode. The positive wool feature at 1485 nm can be assigned to first order N-H/O-H stretching overtones [24]. The negative feature at 1375 nm corresponds well with a feature in the first derivative polymer spectra and can be assigned to the combination of the C-H bending mode and the first order overtone of the C-H stretching mode. These features are responsible for the separation of the wool and contaminated wool spectra.
Editor’s Note: We will be covering chemometric procedures in a future edition this year.
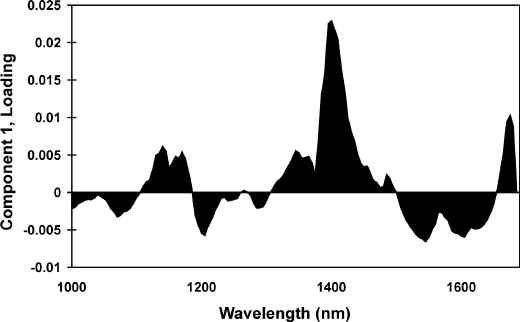
Figure 5. PC1 loadings plot for the 1st derivative, 1000 to 1700 nm region.
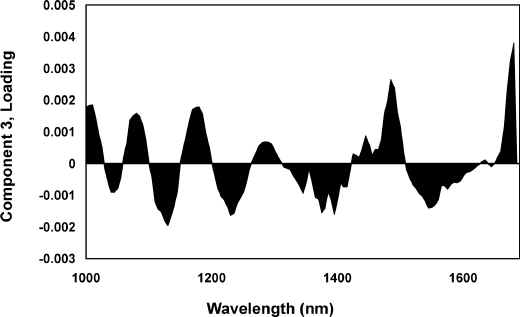
Figure 6. PC3 loadings plot for the 1st derivative, 1000 to 1700 nm region.
Dynamic Experiments
The processing of wool involves wool moving on a conveyor belt after scouring and drying. It was, therefore of interest to determine if the polymer contaminants could be detected in a moving bed of scoured wool. Using the experimental setup described above, data sets consisting of 60 spectra each were obtained from the aluminum sheet covered with both loose wool and polymer contaminated loose wool. The sheet was pulled past the NIR beam at a number of speeds between 1 and 5 metres per minute.
The best PCA results were again obtained using first derivative data from the 1000 to 1700 nm region. For this model 4 PCs account for only 92.5% of the total variance (PC1=53.3%, PC2=31.9%, PC3=4.5%, PC4=2.8%). The PC1 versus PC2 scores-scores plot is shown as Figure 7. This plot exhibits one main pure wool cluster along with a number of outliers. All of the outliers can be associated with the polymer contaminants situated in the bed of scoured wool. As a number of spectra can be collected between the time a piece of contaminant enters and leaves the beam it is thus possible to follow its progress using the scores-scores plot. As it enters the beam it appears as a data point near the edge of the main cluster. When it is directly under the beam it is at its greatest distance from the cluster. As the contaminant leaves the beam the data point merges back into the cluster. This is illustrated by the data points labeled 12 through 20 in Figure 7. The larger the piece of contaminant the further it moves away from the pure wool cluster indicating that the variance is greater.
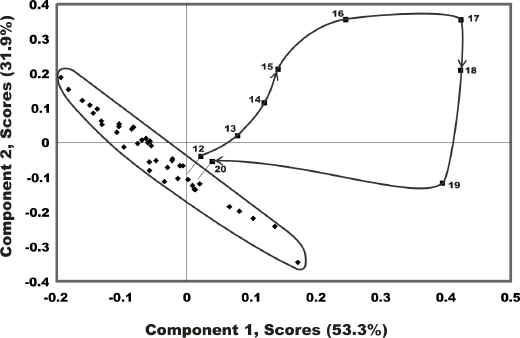
Figure 7. The PC1 versus PC2 scores-scores plot obtained using 1st derivative data from the 1000 to 1700 nm region. The data set was collected while the wool was moving under the NIR beam. The data points labeled 12 through 20 represent a single piece of polymeric contaminant as it passes under the NIR beam.
Summary
In this paper we have presented a brief overview of the principals of near infrared spectroscopy and principal components analysis using a textile based application for illustrative purposes. The coupling of these techniques has become more and more common in industrial situations as faster data collection and processing and more robust instrumentation has become available. The potential applications are only limited by ones imagination.
Acknowledgment
The authors recognize the support of the Australian Wool growers and Australian Government who fund research and development through The Woolmark Company. They are also grateful to Perten Instruments for the loan of the DA 7000 spectrometer.
References
- J. S. Church and A. L. Woodhead, “Rapid Regain Measurement in the Mill”, Proceedings of Top-Tech ’96, Geelong, CSIRO, 1996. p. 376-379.
- Handbook of Near-Infrared Analysis, D. A. Burns and E. W. Ciurczak Eds. Practical Spectroscopy Series, Vol. 13, Marcel Dekker, Inc., New York, 1992. See for example chapters 17 and 18.
- S. Ghosh, “At-Line and On-Line Characterization of Textile Products Using the NIR Spectroscopy Method”, Leaping Ahead with Near Infrared Spectroscopy, G. D. Batten, P. C. Flinn, L. A. Welsh and A. B. Blakeney Eds. 1994, pp. 450-460.
- H. Hoyer, Process Control and Quality, 9 (1997) 143.
- G. Monin, Meat Science, 49 (1998) S231.
- P. Dallin, Process Control and Quality, 9 (1997) 167.
- K. Stromberg, Process Control and Quality, 9 (1997) 179.
- M. Majara, F.M. Mochaba, E.S.C. Oconnorcox, B.C. Axcell, A. Alexander, Journal of the Institute of Brewing, 104 (1998) 143.
- Wham Vandenbroek, D. Wienke, W.J. Melssen, L.M.C. Buydens, Analytica Chimica Acta, 361 (1998) 161.
- P.H. Hindle, Process Control and Quality, 9 (1997) 105.
- G. N. Heintze, CSIRO Division of Wool Technology Report No. G73 (1992).
- J. S. Church, J. A. O’Neill and A. L. Woodhead, Appl. Spectrosc., 52(1998) 1039.
- J. S. Church and J. A. O’Neill, Vibrational Spectroscopy, In Press.
- W. Kaye, Spectrochim. Acta 6, (1954) 257.
- W. Kaye, Spectrochim. Acta 7, (1955) 181.
- Introduction to Infrared and Raman Spectroscopy, 3rd ed, N. B. Colthup, L. H. Daly and S. E. Wiberley, Academic Press, Inc., London, 1990.
- M.F. Dexaux, D. Bertrand, P. Robert, M. Qannari, Appl.Spectrosc. 42(1988) 1015.
- M.F. Dexaux, D. Bertrand, P. Robert, M. Qannari, Appl.Spectrosc. 42(1988) 1020.
- C. Gilbert , S. Kokot and U. Meyer, Appl. Spectrosc. 47 (1993) 741.
- A.L. Woodhead, F.J. Harrigan, J.S. Church, Vibrational Spectroscopy 15(1997) 179.
- S. Kokot, Nguyen Anh Tuan, L. Rintoul, Appl. Spectrosc. 51 (1997) 387.
- C. Gilbert and S. Kokot, Vibrational Spectroscopy, 9 (1995) 161.
- G. Downey, P. Robert, D. Bestrand, P.M. Kelly, Appl. Spectrosc. 44(1990) 150.
- J. Wang, M. G. Sowa, M. K. Ahmed and H. H. Mantsch, J. Phys. Chem. (1994) 4748.
Received 13th January 1999, received in revised format 27th January 1999, accepted 27th January 1999