What is Raman Spectroscopy?
![]()
6.What Is Raman Spectroscopy?
Patrick Hendra
Every year I give several lectures on FT-Raman and its applications in chemistry, pharmaceuticals and materials, and invariably find I have to start by “reminding” my audience what the Raman effect is and how it differs from infrared. It occurred to me that some readers might find this bit of my talk useful, so here goes. This account is quick and nasty and is NOT intended for the expert!
7. If you take a compound and run its infrared, then its Raman spectrum, you notice that the results are very different. This seems a little odd since both are measuring the same thing – the vibrational behaviour of the sample.
Some vibrations give rise to a change in dipole as they contort and this means that they can resonate with electromagnetic radiation of the same frequency. The vibrational frequencies of most molecules are similar to those of radiation in the mid-infrared. Absorption thereof transfers energy into the molecule causing it to vibrate more violently (the amplitude increases) and so we see our familiar infrared spectrum.
Other vibrations give rise to a change in polarizability as the molecule vibrates and it is these that give rise to Raman scatter. What on earth is the polarizability, you ask! Now molecules consist of a nuclear structure surrounded by a complex field or cloud of electrons. Application of a potential field causes the electrons (not the nuclei) to ebb and flow so that they are slightly concentrated towards the + and away from the – of the applied field. The ease with which they respond to a given field is described as the polarizability. If the polarizability changes as the molecule vibrates – Raman bands!
8. So – what is the Raman experiment? If you inject monochromatic radiation at frequency vo or wavelength λo into a sample and look at the light scattered off it – surprise, surprise, it seems to be at the same wavelength as the source. Careful inspection however reveals that in addition to this so-called elastic scatter, a tiny proportion is shifted and it is this shifted radiation that is named after Sir C.V.Raman, the Indian scientist who discovered it in 1928.
9. Let us say our molecule vibrates at 500cm-1, i.e. vvib=500cm-1 and further the vibration causes a change in polarizability as it proceeds. We plan to use a green laser as a source operating at 500nm wavelength ≡ 20,000cm-1. Then the scattered light will be composed of a component at 20,000cm-1 plus two incredibly weak side-bands at 19,500 and another at 20,500cm-1, i.e. at vo ± vvib. We see the experiment below in Fig.1 with correct frequencies filled in.
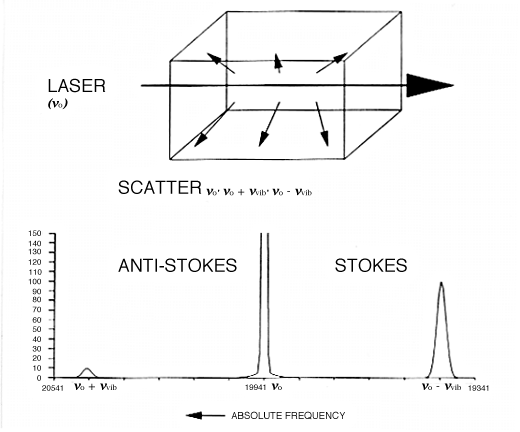
Figure 1: when an intense monochromatic light source of frequency vo irradiates a sample, the light that is scattered contains frequency components vo’ vo + vvib’ and vo – vvib. In the case of liquid chlorine the Stokes and anti-Stokes Raman bands are shifted by 505cm-1 from the excitation frequency. If the spectrum is excited with an argon ion laser vo = 19436cm-1, vo + vvib =19941cm-1 (anti-Stokes) and vo – vvib = 18931cm-1 (Stokes). The Stokes: anti-Stokes ratio is approximately 10:1.
10. So, we have two methods of looking at molecular vibrations and since they originate in different phenomena, there really is no reason why the spectra should look similar. In fact, in centrosymmetric molecules modes give rise to either infrared OR Raman features but not both. Also strong infrared absorptions appear usually as weak Raman ones and vice-versa. In Fig.2 you will see a good demonstration – the infrared and Raman spectra of styrene butadiene rubber.
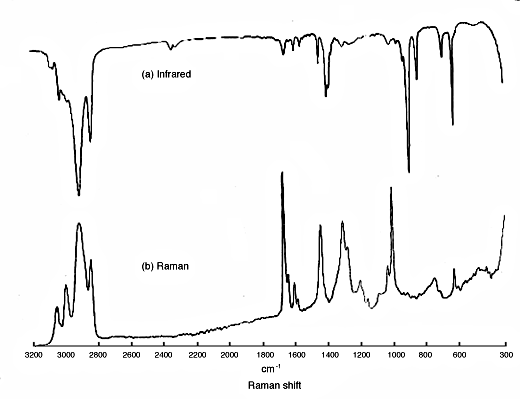
Figure 2: the infrared and Raman spectrum of styrene/butadiene rubber.
11. Now – presentation. The infrared spectra are shown as absorbance vs frequency in wavenumbers but the Raman presentation is very different. The vertical axis reflects the intensity of Raman scatter and the horizontal axis – the frequency SHIFT in wavenumbers. Why shift? When you record a Raman spectrum, you can use any source you like from the UV to the near infrared (certainly 200nm wavelength → 1.3µ). As a result υo can vary over a huge range. We are interested in the vibrational frequency, so we use as a scale the Raman frequency vs the laser frequency i.e. vo ± vvib – vo = ± vvib. Presented this way, the infrared and Raman data can be presented on one diagram.
12. Let’s go back a step and look again at the origin of Raman scatter. When a photon of source radiation hits a vibrating molecule it polarizes it instantaneously, raising its energy by vo. The effect of the interaction is endothermic BUT the excited state is not real – there are no energy levels at the excited condition. As a result we describe these states as ‘virtual’. Leaving out all the jargon – the source photon interacts with the molecule, raising its energy, and the energy is immediately lost again by scattering. The idea is shown in Fig.3.
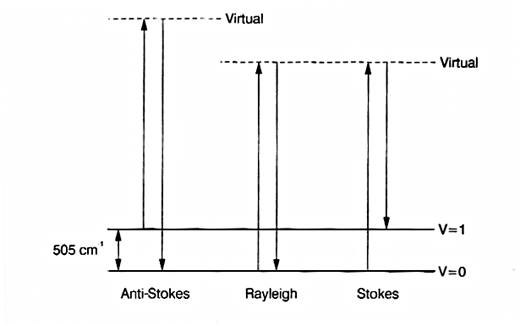
Figure 3: an energy level diagram showing the origin of the Stokes and anti-Stokes bands in the Raman spectrum of liquid chlorine.
13. There are two points to note – by far the most important mechanism produces scatter at υo. A tiny proportion involves a change in the vibrational energy as scatter occurs, yielding Raman bands. Now you will note that the scatter at υo + vvib starts for the vibrationally excited state but that at υo – vvibstarts its course at the ground state. The population of the excited vibrational state is less than that of the ground state.
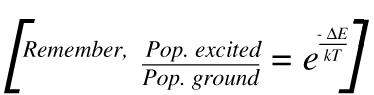
As a result, the υo + vvib (known in the trade as the anti-Stokes lines) are weaker than the red shifted spectrum where they appear at υo – vvib (the Stokes lines). This point is clear in Fig.1. The ratio of the intensities of equivalent pairs of lines I anti-Stokes/I Stokes falls as the vibrational frequency increases and decreases with temperature. This point is shown in Fig.4.
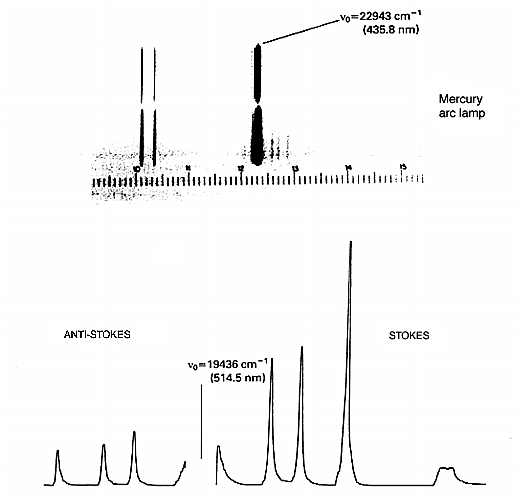
Figure 4: Raman spectrum of carbon tetrachloride. Note the anti-Stokes are always weaker than their Stokes relatives. As Δv.
14. One of the principal methods of interpreting infrared spectra is to use the concept of group frequencies. Thus, vc=o appears around 1700cm-1, vCH aliphatic near 2930 and vOH at 3200-3500cm-1. These correlations occur because compounds containing these fragments reliably produce strong infrared features we can all identify. We have already agreed that strong infrared features are characteristically weak in the Raman effect, so it stands to reason that the infrared group frequencies cannot apply in the Raman and vice versa.
To demonstrate this point – consider the C=C stretching motion: this is not a useful reliable infrared correlation – if the unsaturated group lies near the centre of a molecule, you do not see an absorption at all! On the other hand, vC=C is a really strong Raman feature near 1670cm-1. The frequency enables you to identify cis from trans from vinyl groups.
15. To pick up some of these points, below I show an infrared and a Raman spectrum together and identify some of the group frequencies.
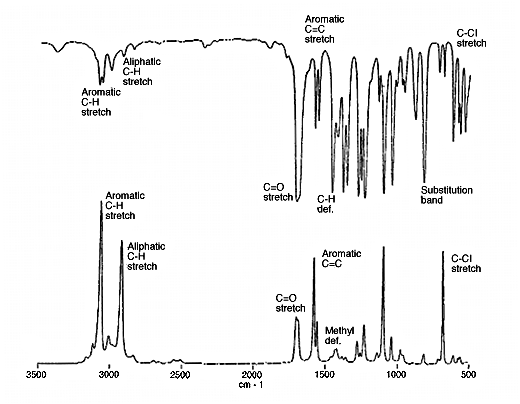
Figure 5a: The infrared and Raman spectra of 2.5-Dichloroacetophenone.
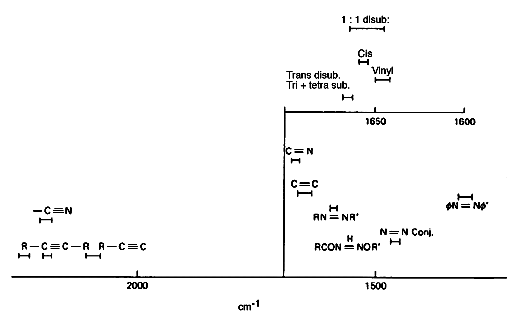
Figure 5b: Frequencies of Raman bands due to unstaurated groups.
16. So, to conclude this introduction – if you want to get the whole vibrational picture, you should run both the i.r. and the Raman and not rely on one spectrum on its own.
When you listen to a tape on your Walkman, you expect to hear the sound in stereo. The music is the same in both ears of course. IR+R = Stereo vibrational spectroscopy!
REF: Int. J. Vib. Spect., [www.irdg.org/ijvs] 1, 5, 6-16 (1998)