A novel method of monitoring polymerisation reactions using Raman Spectroscopy
![]()
Andy Brookes
Department of Chemistry, University of Southampton, Southampton, Hampshire. U.K. SO17 1BJ
45. Abstract
| A novel cell has been constructed to enable polymerisation reactions to be monitored in situ using Raman spectroscopy. The cell is described and several applications discussed. |
46. Introduction
The measurement of reaction kinetics is of fundamental importance. In the case of polymerisation reactions, numerous methods have been developed to follow the reaction as it occurs, in particular dilatometry. This involves measuring the volume of the polymer (or solution) which will change as the monomer reacts due to the long van der Waals forces between monomers being replaced by shorter covalent bonds in the polymer. Refractive index measurements can also be used since these vary almost linearly with conversion.
NMR and elemental analysis have been used to monitor polymerisation reactions. These techniques involve stopping the reaction working up the mixture, removing the pure polymer produced and measuring the yield. Working up may introduce errors into the analysis.
The monitoring of polymerisation reactions is an area where Raman spectroscopy may have advantages over other techniques. It is non-destructive and non-invasive, no work up is required to obtain useful data, spectra can be obtained through the glass walls of reaction vessels and fibre optic probes can be used to provide remote detection. Several of these advantages are discussed in a review on the application of Raman spectroscopy to polymers and polymerisation processes[1]. Raman is a scattering process and so allows the monitoring of the bulk sample as opposed to infra-red where spectra are obtained by transmission through thin films. Thin films can give rise to skin effects which result in misleading kinetic data[2].
47. Several authors have used Raman spectroscopy to study polymerisation processes on and off line. Some early work was done on the homopolymerisation of styrene and methyl methacrylate[2], followed by the copolymerisation of these two monomers[3]. Work has also been done on the copolymers of methyl methacrylate and 1,3-butadiene, the curing of epoxy resins and the thermal degradation of polysulphoxides into polyacetylenes has been explored. This indicates that there are a large number of monomers which are applicable to Raman study.
Several authors have used small volumes of reactants contained within a heating element in the sample area[4]. Others have used large volumes, contained outside the spectrometer and pumped the reacting mixture through a flowcell inside the sample area[5]. Until recently[6] no one has been able to monitor large scale ( more than approximately 1 ml) polymerisation reactions completely inside the instrument.
This paper describes, in detail, the latest kinetics cell designed at Southampton, fig.1. Also discussed are several reactions carried out using the cell. Firstly, the homopolymerisation of styrene in benzene. This was chosen as a model as it is well understood with many studies of this reaction in the literature[7]. Secondly, the emulsion polymerisation of vinyl acetate as described in Ref.5. This provides a comparison with the most recent of the large scale Raman polymerisation cells. The copolymerisation of styrene and vinyl imidazole[6] is also discussed. Large data sets were obtained and the analysis was performed using a software package called SIMPLISMA.
48. Experimental
The metal cell is 150 mm high and stands on a base measuring 128 x 128 mm. This allows the cell to fit easily into the sample compartment of a PE 2000 and other Raman spectrometers. The base can be fixed to the xyz sample stage to allow the cell to be manoeuvred in the laser beam and to have its position fixed reproducibly. All the metal components (excluding the stirrer) are of heavily black anodised aluminium. This provides an inert surface even at elevated temperatures and is also non-reflecting. The cell has a capacity of 40 ml.
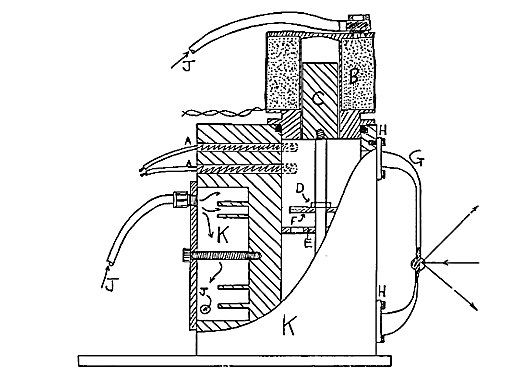
Figure 1: Diagram of kinetics cell
Heating is supplied by two 40 watt low voltage cartridge heaters A monitored by a type T thermocouple. These are capable of maintaining temperatures up to 140°C ± 0.25°C when used with a sophisticated differential proportional controller such as the model D1 Ventacon power supply. To minimise electrical noise generation a D.C. supply is used which operates through transistorised switching.
Energy from the power supply is provided to stir the mixture by powering a solenoid which also doubles as the lid, B. It is powered by a 12V supply and draws 1.5 amps but operates at approximately 1Hz. with a duty cycle of 10%. This ensures heating of the cell and sample compartment by the solenoid is kept as low as possible. Leakage between the body and the solenoid/lid is prevented using a rubber O-ring sitting in an open groove. This is easily replaced if it becomes swollen by solvents. A nickel plated iron rod C makes up the core of the solenoid.
49.A valve is fitted to C and is made of PTFE discs. This ensures the flow of the solution through the side arm. A PTFE disc F sits upon disc E, so that it covers the holes in the bottom disc. As the discs are raised they are pushed together. The solution is then forced through the sidearm. When the pump moves down the top disc is forced against the restraining nut D, allowing solution to pass upward through the valve. The system operates well even when the solution is viscous.
A glass sidearm loop G is incorporated to allow semi continuous Raman analysis of the cellsê contents. A seal is maintained between the glass side arm and the metal body of the cell using PTFE tape between the metal and glass faces. Anodised aluminium rings H are tightly fastened over these to complete the seal.
A bulb I is included in the loop at a point suitable for Raman analysis. The reverse outer face of the bulb is externally mirrored. This reflects more of the scattered radiation towards the collection lens and roughly doubles the signal obtained. This principle is used by Perkin Elmer in their FT-Raman liquid sampling system.
Compressed air J is continuously passed through the solenoid to reduce any heating effect it may have. It is also passed through the body of the cell K to provide a constant thermal load. During non-exothermic reactions it is not essential but does provide essential cooling during exothermic reactions.
The compressed air from the solenoid and the cell body is allowed to vent into the sample compartment. This to some extent minimises heating of the air surrounding the cell by circulating the air within the sample area and of course encouraging it to escape.
50.The reasoning behind the cell design (which has evolved over some time) is as follows. Cells involving an external reactor, pump and flow cell are unattractive because the temperature of the sample is hard to control. The flow lines must be small and flexible so that they are small enough to pass through the light trap into the sample chamber making the study of viscous solutions problematic. Leakage or failure of the tubing has inevitable disastrous consequences to expensive equipment. Our original approach was to use a cell similar to that described here but made of glass. It was heated and cooled by circulating water but here tube failure and consequent leakage of large quantities of water into the sample chamber would be disastrous.
The new design uses only compressed air and electrical power, hence only cables and a single flexible tube pass into the sample chamber. Thus the risk of component failure and subsequent damage to the instrument is greatly reduced.
The spectrometer used was a Perkin Elmer NIR FT-Raman 2000 with a Nd3+:YAG laser operating at 1064 nm, a quartz beamsplitter and an InGaAs detector. The instrument was operated using Perkin Elmer Spectrum software.
All chemicals were supplied by Aldrich Ltd. They were treated to remove any inhibitors and stored at -5°C until use.
51. RESULTS
Homopolymerisation of Styrene
Styrene runs were carried out over ~4 hrs. Over this period the reactions went to approximately 55% conversion, fig. 2.
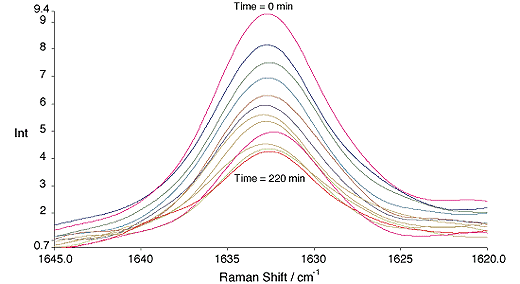
Fig.2, Change in the C=C band of styrene with time. All spectra are recorded at 20 min. intervals. 300mW, 4cm-1, 64 scans.
52.Three chemometric methods were used to analyse the amount of styrene that had been polymerised and to give an indication of the errors which are involved when using each particular method; peak height, peak area and finally deconvolution and estimation of the area under the whole spectrum.
Deconvolution and estimation of the area was difficult as the reactant and product had similar spectra. Peak area was much more sensitive to variations in the baseline than peak height. Therefore, only peak height will be used in the analysis of vinyl acetate polymerisation as this has been shown to be the most accurate method.
% conversion was then plotted against time, fig. 3. It can be seen that these results, obtained at 70°C compare reasonably with those of Gulari, McKeigue and Ng obtained at 80°C.
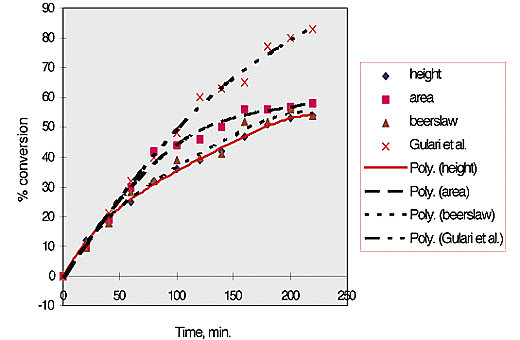
Fig.3, % conversion versus time for styrene polymerisation, analysed using selected analytical methods.
It should be noted that the size of the symbols represents the size of the error in the measurement on the Y axis, ±2.5 %.
The initial rate of conversion of monomer to polymer is 22%/hr. This continues for approximately 80 min. The rate then drops to 10%/hr for the next 80 min. and falls again to 5%/hr for the final 80 min.
53.Emulsion Polymerisation of Vinyl Acetate
Vinyl acetate polymerisations were carried out over ~60 min. During this time the reaction went to approximately 90% completion, fig. 4.
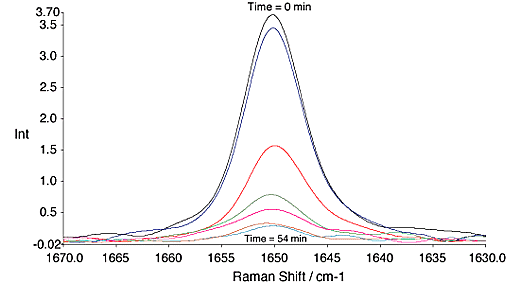
Fig.4, Change in the C=C band of vinyl acetate with time. All spectra are at 9 min. intervals. 300mW, 4cm-1, 64 scans.
54.% conversion was then plotted against time, fig. 5. It can be seen that these results differ somewhat from those of Ozpozan, Schrader and Keller[5].
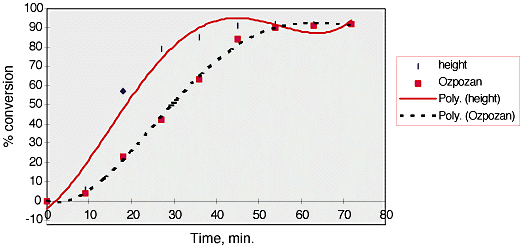
Fig.5, % conversion versus time for vinyl acetate polymerisation analysed using peak height.
Again the size of the symbols represent an estimate of the errors on the Y axis, ±2.5 %.
55.Three phases can clearly be observed in the polymerisation process. The first phase, 0 – 10 min., is the initiation of the reaction. The rate is modest due to the formation of radicals which then go on to propagate the reaction.
The second phase of the polymerisation, 10-30 min., is the propagation of the reaction. This is indicated by the linear acceleration of the reaction in this region. The slope of the curve in this region is approximately 4 %/min.
As the viscosity of the solution increases and the quantity of remaining monomer decreases the rate slows, 30 – 54 min. The reaction reaches 90% conversion in 50 min. which agrees well with Ozpozans’ results.
As can be seen our reaction (and repeats thereof) proceeds more quickly than that of Ozpozan ( we get a slope of ~4%/min whereas Ozpozan has a slope of ~2%/min). We are unsure as to why our reaction proceeds quicker. It could possibly be due to cooling of their mixture as it passes through the tubing into and out of the sample chamber but if there is an effect it would probably be very small.
Another option is that we have inadiquate stirring. This may isolate the reaction mixture in the side arm, possibly causing an increased rate of reaction at the sampling point. This is thought unlikely as when spectra are not being taken the mixture can be seen to pass through the side arm.
Any exothermisity would have been monitored by the thermocouple and heat removed using the compressed air supply.
56. Copolymerisation of styrene and vinyl imidazole
Copolymerisation reactions were carried out over ~6 hrs. More data points were collected in the first 2 hrs. when the reaction would be expected to be at its fastest.
Large amounts of data can be generated for each individual run, fig.6. Spectra can be taken every three minutes and plots of peak height versus time can be produced for both styrene, fig.7, and vinyl imidazole, fig.8. To obtain this data the SIMPLISMA program was used. This is a tool for self modelling mixture analysis, resolving mixture data into pure component spectra and concentrations.
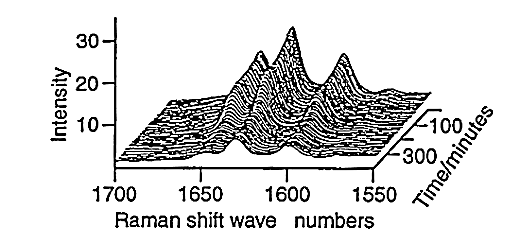
Fig.6, Raman spectra against time for styrene-vinyl imidazole copolymerisation.
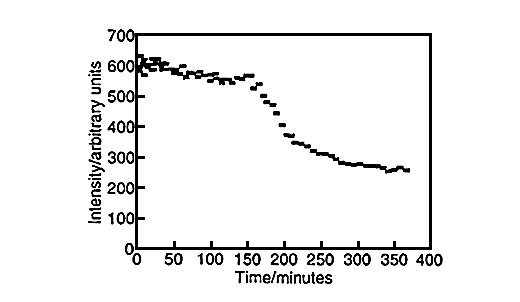
Fig.7, Concentration profile of styrene. Produced using data generated by SIMPLISMA.
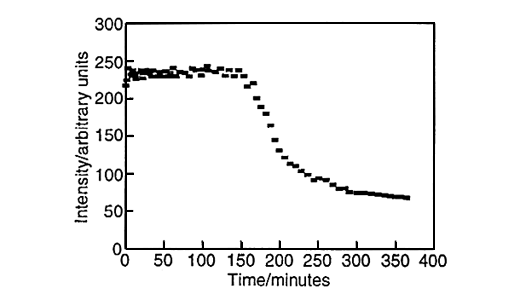
Fig.8, Concentration profile of vinyl imidazole. Produced using data generated by SIMPLISMA.
57. Conclusion
There is no doubt that in situ cells of this type offer a practical method of following reactions without reaction stopping, aliquoting or work-up. They are therefore convenient and labour efficient.
Clearly the system has limitations. To give an acceptable signal to noise ratio spectra are acquired for ~3mins. It is therefore possible to take a spectrum every 3.5 mins. This obviously limits the reactions applicable to study. Until recently we have been unable to look at very fast reactions. When the cell was used with a Reneshaw Ramascope the data acquisition time was reduced to 25 secs. This allows us to look at much faster processes with far more reactive materials e.g. the reaction of HBr across the double bond of vinyl acetate.
The exothermicity of a reaction such as this can be dealt with by the cell. If a reaction is very exothermic the temperature can rise out of control and of course increase the reaction rate.
As shown in the vinyl acetate polymerisation, aqueous solutions are ideal for this type of work as water gives no Raman spectrum. We are restricted to fairly concentrated solutions in order to provide strong enough spectra for analysis.
Very high viscosity can also cause a problem with the stirrer. This results in some of the solution being trapped in the side-arm and spectra being taken from a static, unmixed solution.
The cell is constantly undergoing development in order to overcome these problems and achieve better performance.
Acknowledgements
Alan Strawn and Chris Nicholas at Eastman Kodak Ltd. Harrow.
Prof.P.J. Hendra for all the help with cell design and maintenance.
The kinetics cell discussed in this paper is shortly to be offered as a commercial product by Ventacon Winchester Ltd. Fax:44 1962 776390
References
1. H.G.M. Edwards, A.F. Johnson and I.R. Lewis, J. of Raman Spec. 1993, 24, 475-483
2. E. Gulari, K. Mckeigue and K.Y.S. Ng, Macromolecules, 1984, 17, 1822
3. H.J. Bowley, I.S. Biggin and D.L. Gerrard, Time Resolved Vibrational Spectroscopy, A. Laverbreau and M. Stockberger(Eds.), Vol.4, p.194. Springer Proc. Phys. 1985
4. J. Clarkson, S.M. Mason and K.P.J. Williams, Spectrochim. Acta, 1991, 49A, 1345
5. T. Ozpozan, B. Schrader and S. Keller, Spectrochim. Acta, 1997, 53A, 1 – 7
6. J. Haigh, A. Brookes, P.J. Hendra, A. Strawn, C. Nicholas and M. Purbrick, Spectrochim. Acta. 1997, 53A, 9 – 19
7. K. Witke and W. Kimmer, Plaste und Kautschuk, 1976, 23, 11, 799
* With appropriate modifications the cell can be mounted in any FT-Raman instrument where it is possible to introduce leads and tubes into the closed sample area. The cell has also been used on the Reneshaw Ramascope fitted with macropoint accessory.
REF: Int. J. Vib. Spect., [www.irdg.org/ijvs] 1, 5, 44-57 (1998)