New Progress in Surface Raman Spectroscopy of Platinum Electrode Surfaces
![]()
B. Ren, J. L. Yao, X.Q. Li, W.B. Cai, B.W. Mao and Z.Q. Tian*
State Key Laboratory for Physical Chemistry of Solid Surfaces, Institute of Physical Chemistry and Department of Chemistry, Xiamen University, Xiamen 361005, China
Abstract
By using a confocal microprobe Raman spectroscopy in conjunction with a unique electrochemical surface pretreatment procedure for platinum (Pt) electrodes, we are able to extend the detailed surface Raman studies, for the first time, to the adsorption behavior at bare Pt electrodes in a wide potential region. The surface Raman spectra of pyridine, halide ions (Cl– and Br–), redox couple (Fe(CN)63-/4-), have been obtained in the present study.
Introduction
Vibrational spectroscopy is a particularly valuable tool for investigating electrochemical surfaces because, with careful experimental design, information is to be had about the species present in the electrochemical double layer. Of the methods available, Raman spectroscopy looks particularly attractive because one can use near UV, visible or nir sources, the electrolyte and the cell containing it are transparent, and relatively conventional electrochemical cells can be used inside the spectrometer sample area. Further, electrochemical stimuli can be applied in situ. On the other hand, Raman spectroscopy is an insensitive technique and hence the method has made little impact on the surface chemical field. In the absence of some form of resonance or surface enhancement assistance, the differential Raman cross-section (dδ/dΩ)NRS is less than 10-29cm2sr-1 whence a monolayer of an adsorbate will produce less than one photon per second of Raman scatter in a fairly conventional Raman instrument [1].
The discovery of surface enhanced Raman scattering (SERS) changed the situation for Raman spectroscopy. SERS was first observed by Fleischmann, Hendra and co-workers in 1974 [2].
Later, two groups (Jeanmair and van Duyne, and Albrecht and Creighton) [3,4] showed that Raman intensities for pyridine adsorbed to electrochemically roughened Ag electrodes were six orders of magnitude larger than those expected from the known differential Raman cross section for pyridine in solution and hence an approximately 106-fold surface enhancement of Raman scattering was being observed. These findings stimulated worldwide experimental and theoretical research into SERS. Several mechanisms to explain the effect have been proposed. Among various propositions, two kinds of models have appeared, classified as “electromagnetic” (EM) and “chemical” or “charge transfer” (CT) enhancement. These have been widely accepted as the two main contributions to the giant surface enhancement [5-8]. In the past two decades, with its relative simplicity and considerable sensitivity, SERS has provided a wealth of information on our understanding of solid/liquid and solid/gas interfacial structures, but its application has been restricted very largely to noble metals such as Ag, Au and Cu. This is unfortunate because so much electrochemical research concentrates on platinum and other transition element surfaces which are of value as electrocatalysts. Further, conducting polymers are of vital interest. As a consequence, considerable effort has been expended on extending the range of metals accessible to SERS [9-18]. Of all the efforts, perhaps that centred on the Pt electrode is of the utmost importance, in view of its high electrocatalytical activity, its wide polarizable potential window in practical applications, and its value in fundamental studies of interfacial structures and reactions. Historically, one way to extend Raman spectroscopy to the study of the adsorption at Pt electrodes is to coat the metal onto SERS active Au electrodes as an ultrathin film using underpotential deposition (upd) [10], or alternatively electroplating silver islands onto a Pt substrate [11]. Now the EM enhancement created at gold or silver surfaces is relatively long in range hence weak but observable and hence relatively weak SER spectra of adsorbates on Pt have been obtained.
However, it is very difficult in this complex and demanding method to eliminate entirely the possibility that adsorbate bound to the exposed parts of the gold (Ag) or at the boundary between the platinum and gold (Ag) does not contribute to the resultant spectra. The most appropriate, but unfortunately the most difficult, approach would be to obtain surface Raman spectra directly from the bare metal surface without the complexity of the sandwich configuration (adsorbate/transition metal/SERS active metal). This important goal has attracted many groups since the earliest days of surface Raman spectroscopy. In 1977 Cooney et al. observed the C-O stretching band of very low intensity from the system CO adsorbed onto a platinized Pt electrode [14]. After more than ten years, Bilmes et al. reported an interesting approach. They obtained weak SERS signals from pyridine at electrodispersed Pt electrodes by using a unique roughening procedure to obtain a high surface area [15]. Furthermore, with the recent advances in Raman technique using interferometric (FT-Raman) or dispersive instrumentation, including CCD cameras, three groups have attempted to make a relatively straightforward observations of normal (or resonance) Raman signals from organic molecular layers over smooth mechanically polished Pt electrodes. These have included Pettinger et al. [16], Fleischmann et al. [17], and Pemberton et al. [18]. In all of these reports on bare platinum electrodes [14-18] the surface spectra had to be obtained under absolutely optimized conditions or by some sort of difference spectrum method, due to the insufficiency of signal intensity. Unfortunately, in all these cases, the signals were too weak to be investigated as a function of electrode potential. As a consequence, there have been no new reports on work at bare platinum electrodes in the past four years, indicating the extreme difficulty of investigation of the adsorption behaviour of this metal.
Very recently, taking advantage of a confocal Raman microprobe [19], and especially a unique electrochemical oxidation-reduction (ORC) procedure to get an electrochemically activated platinum electrode surface [15], we have been able to extend, for the first time, the detailed surface Raman studies to bare platinum electrodes over a wide potential region (e.g. -1.0 V to +1.4 V vs SCE) and to more general adsorbates such as SCN, CO and hydrogen, where the Raman scattering cross section [20,21] is small.
In this paper we will show, first that the advance in Raman instrumentation, and the pretreatment of electrode surfaces, are both necessary if one is to obtain good quality surface Raman signals. We will do so by comparing the surface spectra for pyridine adsorption on smooth and over roughened Pt electrodes. Then, the spectra of chloride and bromide anion adsorption on Pt will be presented to demonstrate that surface Raman spectroscopy can be successfully applied to detect the adsorbate-metal interaction in the very low frequency region (< 300 cm-1). Finally, we will demonstrate spectra of the redox couple, Fe(CN)63-/4- which serves as an example to illustrate the feasibility of this technique to monitor electrochemical reduction-oxidation processes in general.
Experimental
Raman spectra were obtained using a confocal microprobe Raman system (LabRam I)[22]. The microscope attachment is based on an Olympus BX40 system and uses a 50x long working-length objective (8mm) so that the objective need not be immersed in the electrolyte. A holographic notch filter is incorporated to filter the exciting line and two selected holographic gratings (1800 g/mm and 300 g/mm) were employed. A CCD was used as the detector. The exciting wavelength was 514.5 nm from an Ar+ laser with a power of 30 mW and a spot of ca.3 μm on the surface. The slit and pinhole in the experiments were 100μm and 600μm respectively. With the 1800 g/mm holographic grating, the spectral resolution is 1.5 – 3.0 cm-1 varying with the spectral region. A PAR 173 potentiostat was employed for potential control.
A polycrystalline Pt rod was embedded in a Teflon sheath and used as the working electrode with a geometric surface area of 0.1 cm2. Since proper surface treatment is essential if one is to obtain good quality surface Raman spectra, a roughening procedure was applied to raise the surface Raman signal by increasing the number of adsorptive sites. It was reported by Arvia and co-workers that Pt electrodes of a relatively large surface area can be obtained in acidic electrolytes via the electroreduction of a previously formed thick hydrous Pt oxide layer by applying a repetitive periodic potential [23,24]. In the present study, the Pt electrode was first mechanically polished with 0.3 and then 0.05 μm alumina powder to a mirror finish, followed by ultrasonic cleaning with triply distilled water. The working electrode was then roughened by a procedure similar to that reported by Arvia et al. [23]. In our case, a mild modification was made to obtain a better surface Raman signal, e.g. a square wave of 2 kHz with upper and lower potential of 2.4 V and -0.2 V was applied to the electrode in 0.5 M H2SO4 for 5s to 10 min, depending on the purpose of the study. The potential was then held at -0.2 V until the electroreduction of the surface was completed. The electrode was then subjected to potential cycles between -0.25 V and +1.2 V at 0.5 V/s until all unstable atoms or clusters were removed and a reproducible hydrogen adsorption/desorption peak was obtained, as shown in Fig.1. The surface area can be estimated by the charge quantities of hydrogen adsorption/desorption in the cyclic voltammogram. In most cases, the roughening time was about 10 min, the real surface area is ca. 400 times larger than that of the single crystal. Finally, the electrode was rinsed thoroughly and transferred to the spectroelectrochemical cell for measurement. A large platinum ring served as the counter electrode. All the potentials quoted are versus the saturated calomel electrode (SCE). All the chemicals used were of analytical reagent grade and solutions were prepared using triply distilled water.
Results and discussion
1. Pyridine adsorption at smooth and roughened Pt electrodes
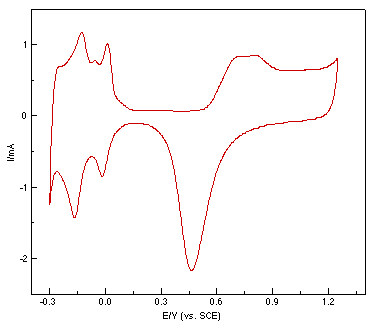
Figure 1: Cyclic voltammogram of a roughened platinum electrode in 0.5 mol l-1 H2SO4.
Fig. 2(a) shows a set of potential dependent Raman spectra of pyridine adsorbed at the smooth Pt electrode. The measurements were carried out in steps, starting from the negative extremity of the potential toward more positive ones, with a spectral acquisition time of 900s for each spectrum. The present result appears to be the first report of potential dependent Raman spectra from smooth Pt electrodes. The experiment has succeeded because of the sensitivity of the confocal microscope – spectrograph – CCD combination. Furthermore, since the confocal microscope has a very good vertical spatial resolution, only a very small amount of the solution is sampled. Therefore, interference from strong Raman signals arising from the bulk solution phase can be effectively eliminated and the different spectrum method for the in-situ IR measurement is unnecessary. In the present study the electrolyte layer is about 1mm thick. However, it can be seen from Fig. 2(a) that a weak band due to the ring breathing mode located at 1004 cm-1 from pyridine in solution, and the broad band due to the HOH bending vibration mode from bulk water do not obstruct the appearance of the surface Raman signal of the adsorbed pyridine at potentials more negative than -0.8V.
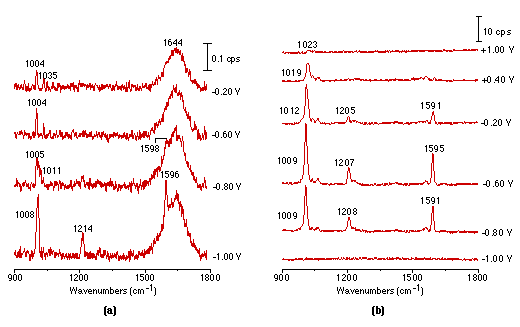
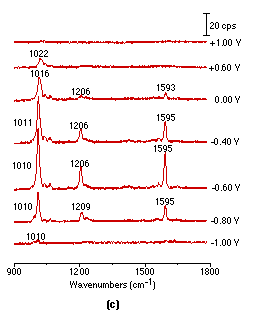
Figure 2: Potential dependent surface Raman spectra of pyridine adsorbed at smooth (a), roughened (b), platinized (c) platinum electrodes.
Solution: 0.01 mol l-1 pyridine + 0.1 mol l-1 KCl.
It should be pointed out that acquisition of the above spectra is really time-consuming. Although it took 900s to obtain each spectrum in Fig. 2(a), a relatively long accumulation time in this type of spectroscopy, the S/N (signal to noise) ratio is still not good enough for practical applications. Obviously, one would wish to persist with smooth surfaces but we decided to try roughened ones, because roughening should enhance the spectral intensity. Using a platinum electrode roughened as described above, we were able to obtain much more intense potential dependent surface Raman spectra of pyridine as shown in Fig. 2(b). Good quality Raman spectra from adsorbed pyridine adsorption could also be obtained from a platinized Pt electrode Fig. 2(c), but plating metallic platinum from a solution containing PtCl42- or even Pb2+ ion may introduce some impurities to the platinized electrodes. One striking difference between Fig. 2(a), Fig. 2(b) and Fig. 2(c) is that the strong pyridine bands over a smooth platinum surface can be observed at -1.0V, but gave extremely weak signals over the roughened surface. Moreover, their potential dependence is also different. These suggest different interactions are occurring between the pyridine and smooth vs roughened surfaces, a point which will be discussed in detail elsewhere [25]. By comparing the surface Raman signal intensity of pyridine over a smooth with that from a roughened metal surface, an enhancement factor of 10-100 was estimated [26]. We emphasize that the platinum electrodes were found to be surprisingly better than Ag, Cu and Au in terms of their stability and reversibility for spectroelectricochemical measurements. The Raman signal could be entirely recovered upon returning to more positive values after a highly negative potential excursion. Moreover, the electrode can be re-used repetitively, provided that the electrode is electrochemically cleaned by cycling in a H2SO4 solution prior to each new experiment. This strongly suggests that the vibrational properties of the adsorbate are reasonably representative of the entire surface, not of some anomaly, an essential requirement for surface Raman spectroscopy to become a general and reliable technique for a wide range of practical applications.
Unfortunately, without the roughening procedure, it is time consuming work to obtain potential dependent surface Raman spectra even for molecules possessing a large Raman cross section and it is obviously impossible to obtain a surface Raman signal from other less favourable molecules. Many of these are of importance and significance, but their small Raman cross section eliminates them from this type of study. It is clear that both the roughening procedure and the advance in Raman instrumentation have contributed to the success of our present work. The following sections are here to demonstrate the applicability of surface Raman spectroscopy to other fundamental or practical systems.
2. Specific adsorption of halide ions on Pt surfaces
The adsorption of halide and pseudohalide ions is one of the most extensively studied systems due to their fundamental importance in surface electrochemistry [27]. They can specifically adsorb on many metals and thus play a significant role in determining the electrode process, such as underpotential deposition (upd) [27]. The vibrational spectroscopic study of their adsorption behaviour has been limited to the SERS-active electrodes [28], because the only accessible vibrational modes of these systems are the bending and stretching of X-M and X-X in the low frequency region, where both the IR and SFG techniques are extremely difficult to operate. Using our newly developed Raman methods, we have successfully obtained vibrational information of metal adsorbate interactions in the low frequency region; lower than 300 cm-1 shift.
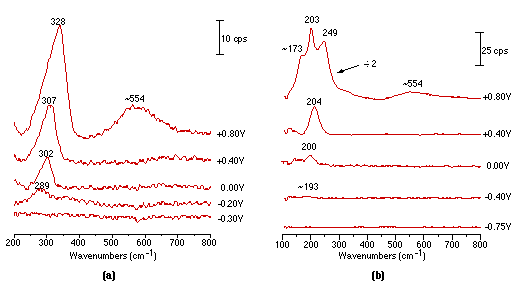
Fig. 3 shows the first Raman spectra obtained over Pt electrode showing the vibration mode of Pt-X. It can be seen from Fig. 3(a) and (b) that, in Cl– and Br– systems, the vibrational bands of Pt-Cl (289 cm-1) and Pt-Br (193 cm-1) are markedly increased in intensity and frequency when positive potentials are used. Significant changes in the frequency and intensity could be detected at the potentials where platinum oxidation occurs,(a band located at ca. 554 cm-1 is attributed to the vibration of Pt-O). For chloride ion, the changes may be due to special interactions between the platinum and the anion, but for bromide ion change is more dramatic. The emergence of bands positioned at 334 cm-1 and at 203 cm-1 and 173 cm-1 may well correspond to the stretching and bending vibration modes of Br-Br and Br3- respectively [29]. The band at 243 cm-1 is probably due to the stretching vibration of Pt-Br influenced by the adsorbed Br2. By comparing Fig. 3(a) and (b), it can be found that the Pt-Br vibrational band can be discerned at -0.4V, whilst in the chloride ion system, Pt-Cl started to appear at -0.2V. On the other hand, the oxidation of Pt was postponed to +0.8V in the bromide system, while in chloride, the potential was -0.5 V.
From the above comparison, one can conclude that the adsorbability of bromine is stronger than that of chlorine.
3. Electrochemical Redox process of Fe(CN)63-/4 at Pt electrodes
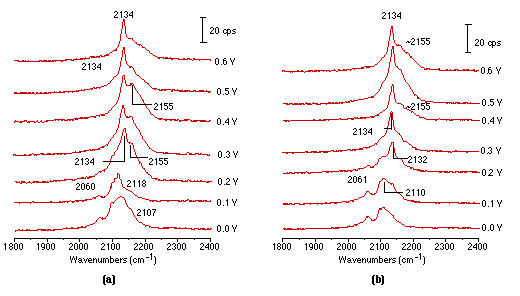
Raman spectroscopy has been used in the past not only for detecting adsorption/desorption processes, but also for monitoring the reduction/oxidation (redox) processes occurring at electrode interfaces, thus revealing something of the electrode kinetics. The Fe(CN)63-/4- redox couple is often considered as a model system for electrode kinetics studies [30-32]. In-situ spectroscopic studies of this couple can provide molecular level information for kinetic studies. There have been some IR studies on this process [33], but the Raman ones have been restricted to Au [34,35] using SERS. No serious surface Raman study at platinum electrodes has been attempted. Recently, we obtained the Raman spectra of Fe(CN)63-/4- during the redox process over roughened platinum electrodes for the first time. In Fig. 4(a) surface Raman spectra illustrate the oxidation process of the Fe(CN)64- ion. It can be seen that two bands positioned near 2054 cm-1 and another at 2110 cm-1 could be detected at potentials from 0 to 100 mV, and due to the stretching vibrations of the CN- ligand correlated to Fe(CN)64-. Further, moving the electrode potential to 200 mV leads to a decrease in intensity of these two ones and they are replaced by two new bands at 2134 cm-1 and near 2155 cm-1 corresponding to the vibrational bands of Fe(CN)63-. This ion produces higher frequencies of vibration than in Fe(CN)64-. The reason is that the δ-bond of the CN ligand in Fe(CN)63- is much stronger than that in Fe(CN)64-. Accordingly, one can reliably assume that at potentials positive of 0.2 volts, Fe(CN)64- is oxidized to Fe(CN)63-. Fig. 4(b) presents Raman spectra obtained from the reduction process of Fe(CN)63- and arises by scanning potentials from 0.6 to 0V. It seems that Fe(CN)63- is adsorbed strongly at the electrode at 0.6V with two bands near 2134 and 2155 cm-1. Moving the electrode potential positively leads to a decrease in intensity of the two bands which are in turn replaced by features close to 2061 and 2110 cm-1, corresponding to vibrational bands of Fe(CN)64-. These appear at potentials positive of 0.2V.
It is necessary to emphasise that in the study of electrochemical redox processes one must be very careful in explaining the Raman results. Two experimental characteristics should be considered for confocal Raman microscopy. Firstly, the solution sample volume is extremely small (about 3x3x3 μm3), much smaller than in conventional Raman spectroscopy. Secondly, an extremely small sampled zone is at the electrode surface where the local concentration of reaction products could be very high and change during the redox process. Therefore, signals from the product in the solution phase could be present in addition to those from the surface. As a consequence, it is very difficult to be absolutely sure in interpreting the detail in adsorption behaviour of the redox couple. A flow cell system and a rotating electrode would be very helpful in sorting out the contributions of the adsorbed species and solution species to the Raman signal, and progress in these areas is hoped for in our laboratory.
Conclusions
The SERS technique, although of immense value, has always been restricted to the copper, silver and gold surfaces. As a result there has been a tendency to use the method for low level analysis, the amount of electrochemical information has been relatively sparse.
By using a unique surface pretreatment procedure in combination with the confocal microscope Raman system, a wealth of information has now been obtained on molecules adsorbed to Pt electrodes, e.g. for pyridine, Cl–, Br– and Fe(CN)63-/4-. Although the structural information discerned from the present results is incomplete and rather qualitative in nature, it clearly demonstrates the virtues of surface Raman spectroscopy in yielding information on surface bonding relating it to the surface coverage, coadsorbates, electrolyte ions and electrolyte potential. It should also be emphasized that the stable and reproducible dispersed Pt surface used in our Raman measurements also offers high electrocatalytic activity and negligible redistribution of the active properties close to those of catalytic materials [22]. Therefore, it must be promising to extend our studies on surface Raman spectroscopy to ultrahigh vacuum environments and to gas/metal interfaces. The progress reported in this article provides a good reason to be optimistic that surface Raman spectroscopy will eventually be used as widely as surface infrared absorption for the study of metals of great practical importance.
Furthermore, we assume with confidence from our results that surface Raman spectroscopy can be extended to investigate adsorption to semiconductors and dielectric films, especially when further improvements in surface pretreatment appear and Raman instrumentation gains sensitivity.
Acknowledgments
This work is financially supported by the Natural Science Foundation of China and the State Education Committee of China.
References
1. B. Pettinger, In: J. Lipkowski and P.N. Ross, Adsorption of Molecules at Metal Electrodes VCH: New York, 1992 p 285.
2. M. Fleischmann, P.J. Hendra and A.J. McQulillian, Chem. Phys. Lett., 26, 163 (1974).
3. D.L. Jeanarie and R.P. Van Duyne, J. Electroanal. Chem., 84, 1 (1977).
4. M.G. Albrecht and J.A. Creighton, J. Am. Chem. Soc., 99, 5215 (1977).
5. R.K. Chang and T.E. Furtak, Surface Enhanced Raman Scattering Plenum Press: New York, 1982.
6. M. Moskovits, Rev. Mod. Phys., 57, 783 (1985).
7. A. Otto, I. Mrozek, H. Grabhorn and W. Akemann, J. Phys. Condens. Matter, 4, 1143 (1992).
8. R.L. Birke and J.R. Lombardi , In: C. Gutierrez and C. Melendress, Spectrosocopic and Diffraction Techniques in Interfacial Electrochemistry, Kluwer Academic, Drodrecht, 1990 p 263
9. M. Fleischmann and Z.Q. Tian, J. Electroanal. Chem., 217, 385 (1987).
10. Y. Zhang and M.J. Weaver, Langmuir, 9, 1397 (1993).
11. H. Furukawa, K. Ajito, M. Takahashi and M. Ito, J. Electroanal. Chem., 280, 415 (1990).
12. L.J. Oblonsky, T.M. Devine, J.W. Ager, S.S. Perry, X.L. Mao and R.E. Russo, J. Electrochem. Soc. 141, 3312 (1994).
13. C.J. Zhong, Z.Q. Tian and Z.W. Tian, Science in China (B), (12), 1233 (1990).
14. R.P. Cooney, M. Fleischmann and P.J. Hendra, J. Chem. Soc. Chem. Commun., 235 (1977).
15. S.A. Bilmes, J.C. Rubim, A. Otto and A.J. Arvia, Chem. Phys. Lett. 159, 89 (1989).
16. B. Pettinger and U. Tiedemann, J. Electroanal. Chem. 228, 219 (1987).
17. M. Fleischmann, D. Sockalingum and M.M. Musiani, Spectrochim. Acta. 46A, 285 (1990).
18. M.A. Bryant and S.L. Loa, J.E. Pemberton, Langmuir, 8, 753 (1992).
19. R. Tabaksblat, R.J. Meier and B.J. Kip, Appl. Spectrosc. 46, 60 (1992).
20. Z. Q. Tian, B. Ren, Y.X. Chen, S.Z. Zou, B.W. Mao , J. Chem. Soc.- Faraday Trans., 92, (1996).
21. Z.Q. Tian, B. Ren, B.W. Mao, J. Phys. Chem., (accepted).
22. S. Sharonov, I. Nabiev, I. Chourpa, A. Feofanov, P. Valisa and M. Manfait, J. Raman Spectrosc. 25, 699 (1994).
23. A.C. Chialvo, W.E. Triaca and A.J. Arvia, J. Electroanal. Chem. 146, 93 (1983).
24. A. Visinitin, W.E. Triaca and A.J. Arvia, J. Electroanal. Chem. 221, 239 (1987).
25. B. Ren, W.B. Cai, B.W. Mao, F.M. Liu and Z.Q. Tian, J. Electroanal. Chem., 415, 175 (1996).
26. W.B. Cai, B. Ren, X.Q. Li, J.L. Yao, B.W. Mao and Z.Q. Tian, (to be submitted to J. Electroanal. Chem.).
27. A.T. Hubbard, Chem. Rev., 88, 633 (1988).
28. P. Gao and M.J. Weaver, J. Phys. Chem., 90, 4051 (1986).
29. K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, 3rd Ed.(Wiley-Inter Science, New York, 1978).
30. R.A. Marcus, J. Phys. Chem., 67, 853 (1963)
31. L.M. Peter, W. Durr, P. Bindra, and H. Gerischer, J. Electroanal. Chem., 71, 31 (1976)
32. A. Wieckowski, M. Szklarczyk, J. Electroanal. Chem., 142, 157 (1982).
33. S. Pons, M. Datta, J.F. McAleer and A.S. Hinman, J. Electroanal. Chem., 160, 369 (1984)
34. R.B. Lowry, J. Raman. Spectrosc., 22, 805 (1991).
35. R.B. Lowry, Spectrochim. Acta, 49A, 831 (1993).
REF: Int. J. Vib. Spect., [www.irdg.org/ijvs] 1, 2, 7 (1996)