Article
![]()
By Patrick Hendra
In a real sense there is hardly any need to write this article, as FT Raman must be just about the most versatile and trivially easy non-destructive analytical procedure ever developed – you can get a spectrum from a screwdriver handle, a pill, a bottle of scotch (unopened) or a lump of cheese, with equal ease. In no case does one need to prepare a sample, just hold it firmly in the machine. Well, this is all very true, but to obtain best spectra there is a little more to it than brute force and ignorance.
All FTs have several features in common – they all illuminate the sample with near i.r. radiation from a Nd3+:YAG laser operating at 1.064 ?. They all collect the scattered light in the reverse direction to illumination – so-called Back Scatter – and they all process the light with a modified F.T.I.R. instrument. The arrangement is shown below:
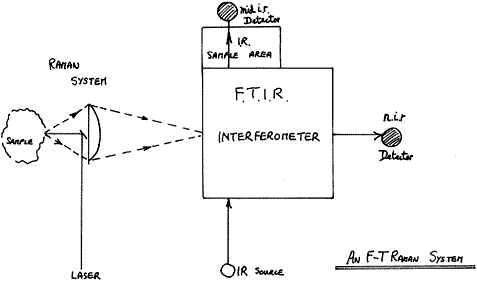
Figure 1
Perhaps the only significant difference between the various competing products is that most collect the light with a lens (or lenses) but some, e.g. Nicolet, use an ellipsoidal mirror. Either way, scatter over a wide angle is collected and passed into the interferometer.
Because the latter has a round hole as its entrance aperture (the so-called Jacquinot Stop), the instrument looks at a relatively large round patch of the sample and it is into this patch that the instrument maker focuses the laser.
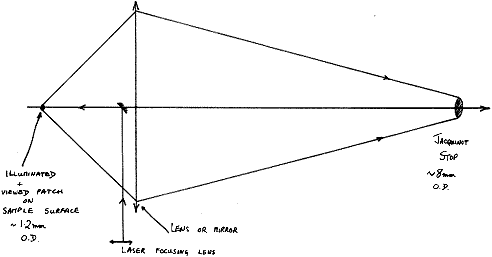
Figure 2
The viewed VOLUME allowing for the depth of focus of the laser is of dimensions a cylinder 0.5-1 mm in diameter and height around 2 mm. Any sample placed inside this space will produce a spectrum.
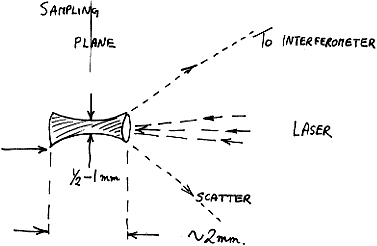
Figure 3
All the commercial instruments have interlocked lids so that the laser radiation, which is invisible anyway, cannot be damaging to the eyes. How do you “find” the sampled volume if the sample holding system is misaligned? Most manufacturers provide some sort of jig, but these tend to be unnecessarily cumbersome. It is a fact that Nd3+:YAG lasers melt black PVC tape, so the trick is to stick a layer of ordinary insulating tape over the sample holder and close the lid. Re-open it and – hey presto! – a tiny hole will be seen where the laser hit the tape.
A liquid can be viewed by containing it in a bottle, vertical tube, ampoule or capillary made of glass or quartz. Since the viewed cylinder has length, the thinner tubes may not be very efficient if viewed normal to their axis.
Clearly, better results would be obtained by viewing down the cylindrical axis but a flat window is then essential at the spectrometer end – all a bit tricky. There are a couple of ruses worth trying if a tube, small bottle or capillary are used. If you place a piece of clean aluminium foil behind the sample, the laser tends to be reflected back into the sample. Similarly, the scattered light leaving the sample away from the spectrometer is not then wasted but rather projected forward into the instrument. A much better technique is to silver the reverse outer surface of the bottle or tube. This is really easy and many recipes for silvering solutions exist. To protect the silver coat we use typists’ correction fluid. Since the silver will then not rub off these, sample bottles/tubes can be used again and again.
Perkin-Elmer have taken this reflection idea further by using either a spherical reflector, inside which lies the sample enclosed in a tiny spherical flask ![]() 5mm in diameter, or in a capillary. Just as good results can be obtained on any machine by blowing a small bulb on the end of a boroscilicate tube and silvering the outer surface over about half of its surface.
5mm in diameter, or in a capillary. Just as good results can be obtained on any machine by blowing a small bulb on the end of a boroscilicate tube and silvering the outer surface over about half of its surface.
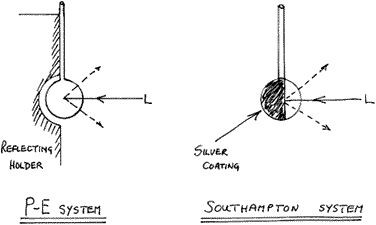
Figure 4
Solids
Lumps – not much you can do with these but beware focusing the laser too deeply inside the bulk. Best results seem to come if the viewed position is at or just beneath the surface. If the sample is glass clear, then move the sample forward and treat more like a liquid.
Powders – the normal procedure is to sample these in a thin glass tube (e.g. a melting point or NMR tube) but we prefer to use a self-supporting compressed pad. In fact, this is the normal procedure at Southampton.
Various cells have been devised
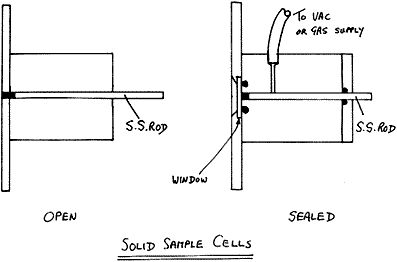
Figure 5
Our standard routine cells are home made 3 mm (1/8″) in diameter but we have made them down to only 0.6 mm in diameter. The quality of the spectrum is not hole size dependent but the fine ones are difficult to fill and tricky to align. One word of caution – for years we made these cells from 1/2″ brass rod because it is easy to drill and relatively corrosion resistant. It seems this was a bad choice – the thin coating of ZnO fluoresces like crazy, so if you miss the centre of the sample you can get a mysterious background. Better to ask your workshop to use aluminium alloy and have it anodized. The anodized surface is almost completely chemically resistant and very hard – ideal.
To grind or not to grind is indeed the question. Some powder specimens give excellent spectra, whatever their state, others improve if ground. The answer to the question is complex but in many but not all cases a bit of grinding can help. We do not actually grind the specimen as such, just move the specimen around as we compress the pad and/or rotate the pressure rod.
Films
A relatively thin, free standing film such as a piece of wrapping material can often give a decent spectrum if it is rolled up and the roll is viewed on axis. The point is that the instrument looks into the sample in some depth. An alternative is to stack layers of the films. An easy and economical way to do this is to use a commercial paper punch (as used for preparing sheets for ring binders) and ask your workshop to make a simple metal holder. Making the bottom disc out of aluminium foil is a good idea.
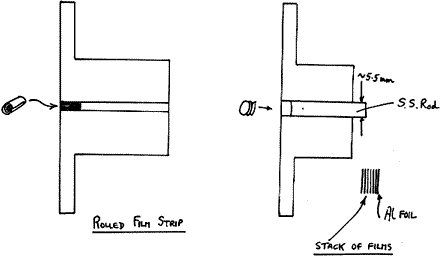
Figure 6
What of films on surfaces? Not a strong point for F-T instruments. We have had luck with 2 ? thick films on reflecting surfaces but if a thin film overlies a dark or rough surface, you will have trouble unless you use a microscope (We will feature infrared and Raman microscopy in the very near future).
There are two distinct problems with F-T Raman operating in back scatter. Both arise because the patch illuminated by the laser is relatively small (compared with the area sampled in an infrared absorption or reflection experiment). The first is burning and the second inhomogeneity, causing the signal strength to vary with position.
Burning
All samples absorb the radiation incident upon them and, as a result, the sample heats. If a laser is focused onto the surface of a solid the brightness of illumination can be very high indeed (3000W cm-2 are ordinary in FT Raman machines). If the sample has restricted thermal conductivity – not at all unusual in powders or polymers, a really significant temperature rise can occur. If heating causes darkening, the consequent heating effect can run away and the sample burns. Obviously, the brightness of illumination must be minimised and the thermal conductivity kept as high as possible. Since, as we have seen, the illuminated area must match the viewed, there is little opportunity to solve the heating problem by fiddling around with the illumination. The only real way to minimise the risk of damage is to keep the laser power down and accept poor signal intensity.
How can you check whether you have a problem? Obviously, if no Raman signal appears and there is a black patch on the sample, you have a problem. There are a few tests you can do. Try lowering the laser power dramatically (say to 50 mW) and attempt to run a spectrum. You may see evidence that the background is rising as you co-add the spectra. If your instrument has a “monitoring or fast low resolution mode”, set up the sample, switch on the laser, and watch the result. The point here is that as the sample heats up, it will emit as a black body (hopefully, not literally!) and you will see the emission as a rising background at high shifts. A typical appearance would be
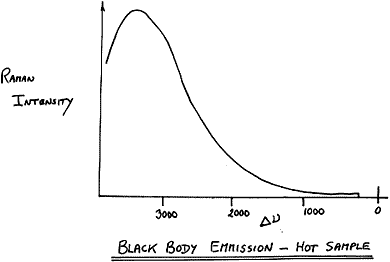
Figure 7
If the sample behaves itself at 50 mW, increase to, say, 100 mW and try again. Emission at high shifts will be evident if the sampled point reaches 120-130°C or higher.
So – how much heating does occur at the surface of a typical sample? It is very hard to measure but Dr. Yvonne West here at Southampton is something of an expert and offers a paper in the Contributed Articles section of this edition which you will certainly find useful.
Above, I pointed out that an alternative way to solve the heating problem is to keep the laser power normal but increase the thermal conductivity to allow the heat produced to diffuse away from the illuminated volume. Dr. Geoff Dent of Zeneca has come up with an excellent idea on this one and Dr. West’s article in the Contributed Articles section covers the subject.
Inhomogeneity
Simple consideration of this problem would lead you to the conclusion that two approaches might be worthwhile – view a larger patch of sample and/or move the sample with respect to the viewed patch so that can occur. There are snags to both solutions. If the optics are altered so that the viewed area is magnified, the solid angle collected by the instrument must be reduced (the f number at the sample will rise). So, the intensity of light collected per unit laser power will fall. Of course, one could increase the laser power to compensate for the fall but in a sense you are then asking for trouble through heating. There is a mitigating factor –
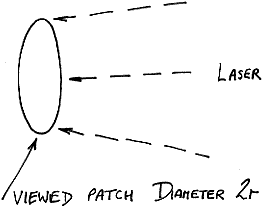
Figure 8
As the diameter 2r of the viewed patch is increased, its area rises of course as r2. As a result, looking at a large area and defocusing the laser before increasing its power can achieve the results we need whilst keeping the brightness of illumination under control.
This is all very well, I hear you say – but does it work? Yes is the answer.
Using a special lens in front of the normal one on a P-E2000R we can achieve around 60% of the signal strength normally achieved and the sample point is moved by about 25mm.
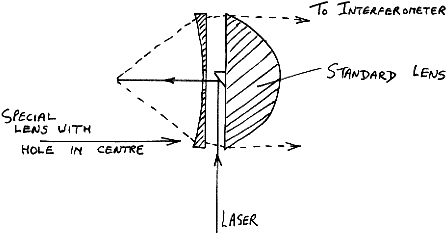
Figure 9
The illuminated and viewed patch is, of course, increased in size (to around 0.5 mm diameter).
As an alternative, how about moving the sample under the beam? You might think that doing this would generate noise in the interferogram which the FT processor would translate to a background. It does, but well outside the audio bandwidth typical of spectra in the 1 – 1.7 ? region if you move the sample slowly.
Rotation of an NMR tube of powder at around 60 rpm is fine and this is frequently used on Nicolet machines, but it would not be a good idea to spin the sample at, for instance, 2000 rpm.
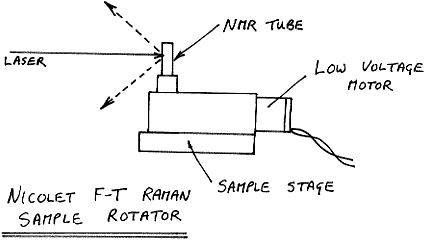
Figure 10
To conclude: F-T Raman is such a simple routine technique that you can get spectra with almost no sampling. On the other hand, it pays to try a few experimental tricks if you want the best results.
Next time, I will discuss heating and cooling, whilst in the future we will highlight infrared and Raman microscopy.
At several places I have described special sample holders, lenses, rotators or other sepcialised pieces of equipment. I am happy to provide full details if you send me a fax on +44-1962-776-390
REF: Int. J. Vib. Spect., [www.irdg.org/ijvs] 1, 1, 4 (1996)