Application of FTIR spectroscopy to probe and improve protein structure in sustained release devices
Kai Griebenow*,
Ingrid Castellanos and Karen G. Carrasquillo
University of Puerto Rico,
Río Piedras Campus,
Department of Chemistry,
PO Box 23346,
San Juan,
PR 00931-3346
* Corresponding author, e-mail griebeno [at] adam.uprr.pr
Introduction
In our contribution published previously in the IJVS (Griebenow et al., 1999) we have extensively described the application of FTIR spectroscopy to characterize dehydration-induced structural changes in proteins. Included were a brief introduction of the theoretical background, practical considerations of how to determine protein secondary structure from IR spectra, and some examples showing what results can be obtained employing this spectroscopic technique. In this overview article we would like to extent the last section of our prior review and focus on relevant examples of recent applications of FTIR spectroscopy in an important area with medical and biotechnological applications. All examples have in common that protein structure is being investigated under conditions not easily approachable by other techniques. Such conditions include (but are not limited to) suspensions of dehydrated protein powders in organic solvents and dispersions of protein powders in polymers. We will outline how FTIR spectroscopy can be employed in a rational optimization of a modern drug delivery system. We named this approach structure-guided protein encapsulation.
Pharmaceutical proteins and their delivery
Many biopharmaceutical drugs, such as peptides and proteins, currently enter or are in clinical trials. This is largely due to recent advances in biotechnology allowing the mass-production of recombinant proteins [1]. Some examples of promising new protein drugs include insulin-like growth factor for the treatment of juvenile-onset insulin-dependent diabetes [2], tumor-derived heat shock proteins for immunotherapy of tumors [3], and antibodies in cancer treatment [4]. The list of already FDA approved recombinant protein pharmaceuticals is steadily growing [5]. Applications of peptide and protein drugs include hormone treatment (e.g., insulin, growth hormone), immuno- and tumor-treatment (interferon, antibodies, interleukins), cardiovascular and thromobolytic treatment (e.g., tissue plasminogen activator, urokinase), and immunization (e.g., immunoglobulins, antitoxins) [6].
However, the use of proteins as biopharmaceuticals is hampered by the fact that patient-convenient routes of administration (e.g., orally) frequently cannot be used due to the susceptibility of proteins to biological degradation pathways, such as proteolysis. Therefore, most proteins are delivered via parenteral routes to the patient [6]. However even when delivered by parenteral (e.g., insulin) or pulmonary routes (e.g., recombinant human deoxyribonuclease I) proteins often possess low biological halftimes and frequent administrations are required. For example, the half-life of interferon alpha, beta, and gamma varies between 25 minutes and 16 hours in humans [6].
A possible and exciting solution to such problems is the controlled delivery of proteins from biocompatible polymers [7-16]. Since it has been established that large molecules including proteins can be delivered slowly and continuously from biocompatible polymers [17], the sustained release of proteins and peptides field from such polymers has grown immensely [18]. These systems provide many advantages over conventional therapeutic approaches, such as intravenous injection. For example, they can be constructed to deliver their active component at a constant rate for prolonged periods, they can target specific tissues, extend the half-life of the drug, and also enhance its in vivo stability [13,18-20]. In addition, patient compliance and comfort, as well as control over blood levels may be improved with the development of sustained release protein injectables, since regular invasive doses can be avoided [18]. Of tremendous value for medical and humanitarian reasons would also be the development of “one shot” immunization [21,22], e.g., by the sustained release of tetanus toxoid [13,19]. Particularly exciting are developments in single-administration vaccines against HIV-1 infection based on the envelope glycoprotein gp120 (currently in clinical trials) [21]. Success in such endeavors would in particular boost efforts by the WHO to achieve high levels of vaccination in developing countries where frequent medical attendance is still a very serious problem [21].
Protein encapsulation for their sustained release
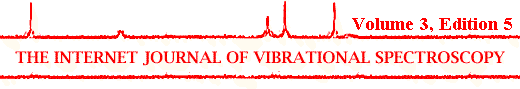
Even though there is a tremendous potential in delivering proteins from biocompatible polymer devices, there are also tremendous problems involved in the encapsulation process. Proteins have a very fragile three-dimensional structure [23-25] and the protein encapsulation process was believed to cause significant protein structural perturbations [18,26]. Such structural perturbations can lead to the formation of irreversible protein aggregates. Aggregate formation not only leads to a loss of expensive pharmaceutical protein, but also can have severe consequences for the patient. First, protein aggregation adds a hard-to-control variable to the precise prediction of the release profile for the drug, which in turn may prove fatal to the patient. Furthermore, protein aggregates can be immunogenic and possibly lead to shock and death of patients. An ultimate goal in the encapsulation of proteins for their sustained release is therefore to assure the preservation of their native three-dimensional structure upon encapsulation and subsequent delivery to avoid formation of protein aggregates. How challenging this requirement is will be explained following a typical protocol used for the encapsulation of proteins. Figure 1 shows the scheme of encapsulation of a protein by the so-called double-emulsion/solvent-evaporation (a.k.a., water-in-oil-in-water or w/o/w) technique. This procedure is by far the most frequently used encapsulation method [18].
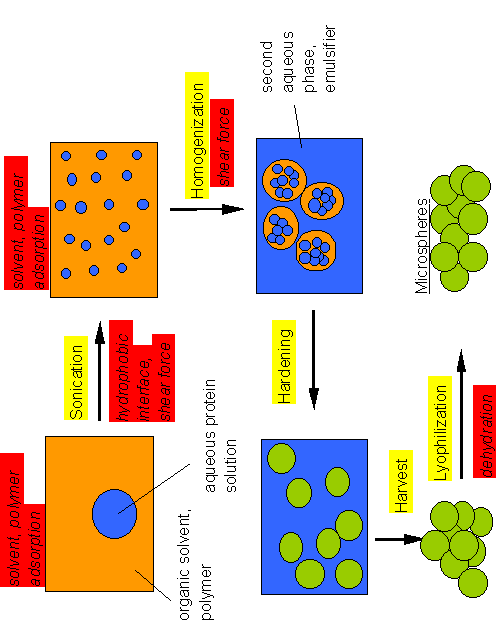
Figure 1. Protein encapsulation by the double-emulsion/solvent-evaporation (w/o/w) technique. The main events likely to denature the proteins are indicated in Italics and are underlayed in red. Modified after Schwendeman et al. (reference 18).
In the w/o/w technique, an aqueous protein solution is introduced into an organic solvent. This solvent is needed to dissolve the hydrophobic polymer [mostly poly(lactic-co-glycolic) acid, PLGA] and is typically methylene chloride because of its excellent properties (good polymer solubility and easy to evaporate) or sometimes ethyl acetate. The first emulsion is formed using a suitable method, e.g., probe sonication or homogenization. During this process a large area of solvent-water interface is formed. It is believed that proteins adsorb into this interface due to their surface activity [18]. Hydrophobic interfaces are known to destabilize the native structure of proteins and this can lead to formation of protein aggregates [18]. In addition, the method of emulsification poses stress upon the protein. After formation of the first emulsion, a second one is formed by introducing the first emulsion into an aqueous solution containing an emulsifier, e.g., polyvinyl alcohol. Again, a homogenization method has to be used. This is next followed by a period of solvent evaporation leading to the hardening of the polymer, typically by stirring for some hours. As a result, the protein solution is trapped into the relative hydrophobic matrix of the PLGA polymer forming small inclusions. Some dehydration is likely to occur due to the solubility of water in the organic solvent (methylene chloride can contain up to 2% of water). Finally, the microspheres formed are collected, washed with distilled water (repeated centrifugation or filtration), followed by dehydration, typically by lyophilization. The final step again imposes significant stress upon the protein because lyophilization causes significant protein structural perturbations (see our previous review in IJVS, Griebenow et al., 1999). In summary, the list of stresses resulting in the destabilization of the three-dimensional structure of proteins is long and includes events such as interfacial-, solvent-, mechanically-, and dehydration-induced protein denaturation. The resulting PLGA-microspheres loaded with protein are shown in Figure 2.
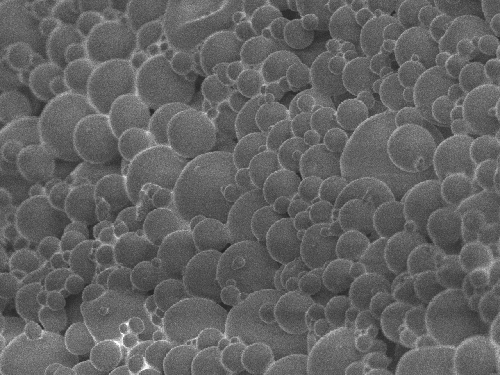
Figure 2. Electron micrograph of protein-containing PLGA-microspheres obtained with a surface scanning electron microscope. The microspheres have an average size of ca. 30 micrometer, but the size distrubution in this preparation is quite heterogeneous.
In the context of this long list of events involved in the encapsulation procedure that could indeed impact upon the structure of proteins and also the reports that abound on the incomplete delivery of proteins from such devices, an urgent goal identified in the literature is to learn how the encapsulation procedure affects protein structure. Until very recently NO information was available on the structure of proteins in such microspheres simply because there was no suitable method that could be employed to study them.
Now, thanks to recent instrumental and conceptual advances in the area of FTIR spectroscopy, (Griebenow et al., 1999) this has changed. As a result, the structure of a protein dispersed as an amorphous powder in a hydrophobic matrix such as PLGA has been determined for the first time by Carrasquillo et al. [27]. In parallel, the secondary structure of lysozyme and BSA was determined for the first time within PLGA microspheres by Fu et al. [28]. The outcome of these and related investigations and their impact on protein formulation for their sustained delivery from biocompatible polymers will be described in this article.
Protein structure in PLGA microspheres created by the w/o/w technique
Fu et al. [28] encapsulated the two model proteins – hen egg-white lysozyme and bovine serum albumin (BSA) in PLGA microspheres following a typical protocol involving the w/o/w technique. The structure of the proteins encapsulated in the microspheres was determined by FTIR spectroscopy and the a-helix content was used as the main structural parameter. For both proteins, encapsulation was found to severely impact the protein secondary structure. These effects can be summarized by focussing on BSA. The a-helix content for BSA plummeted from 54% in aqueous solution to 21% in the PLGA microspheres. Interestingly, this drop was significantly larger than that upon lyophilization (to 31%). One must conclude that the complete encapsulation procedure imposes significantly more stress upon the protein secondary structure than does lyophilization alone, the final step of the encapsulation procedure. Thus, the initial steps in the encapsulation protocol probably contributes significantly to the destabilization of protein structure. In particular, the formation of the first emulsion is suspicious in this context, but protein structural data have not been obtained thus far.
The detrimental structural changes could to some extent be prevented by the use of lyoprotectant trehalose in the encapsulation protocol. The a-helix content of BSA encapsulated in microspheres whilst in the presence of trehalose was 30% and thus significantly higher than in the absence of trehalose. It should be noted, however, that trehalose is only an established lyoprotectant that is efficient in preventing dehydration-induced structural changes [27,29,30]. Its potency in preventing protein structural perturbations caused by other stress factors involved in the encapsulation protocol is unclear. The data indicate that trehalose is obviously not efficient in eliminating all of the various stress factors efficiently in the w/o/w protocol.
Qualitatively, similar conclusions have been reached by Yang et al. [31] for recombinant human growth hormone. Therefore, thus far, a native protein structure has NOT been reported for proteins encapsulated in PLGA by the w/o/w technique.
Conclusions
FTIR spectroscopy has been successfully employed to characterize the secondary structure of proteins in PLGA microspheres obtained by the w/o/w technique. However, significant procedure-induced structural perturbations have been diagnosed under all conditions. Stabilization of the protein secondary structure has been achieved to some extent by employing stabilizing additives, such as trehalose.
Structure-guided protein encapsulation
using alternative techniques
One can surmise from the above results is that there is an urgent need for the development of alternative procedures to encapsulate proteins in hydrophobic polymers. In the following, we will focus primariyl on those aspects of this work that includes protein structural information.
As one possible solution to the problems decided, we have developed recently an approach that we call structure-guided protein encapsulation [27,30]. Herein, FTIR spectroscopy is used to structurally guide the complete encapsulation procedure and any protein structural changes are systematically eradicated. One problem frequently encountered was the fact that the w/o/w technique does not allow us to encapsulate proteins in a unperturbed structure. Too many potential causes for structural changes have been identified and each of these require the development of a stress-specific stabilization method. We felt that the main factor responsible for the difficulties was the employment of proteins in aqueous solution. Under such conditions proteins are flexible – as demonstrated by their typically moderate temperature of denaturation. Most proteins denature reversibly or irreversibly below 100oC [32,33]. In contrast, it is well established that dehydrated protein powders and also suspensions of those in a variety of organic solvents denature at quite high temperatures. For example, suspended protein powders can show catalytic activity for days in neat organic solvents at temperatures far above 100oC [34,35]. This is due to the fact that dehydrated proteins are “rigid” in organic solvents. Thus, even though they can be denatured by the solvents for thermodynamic reasons, due to this increased rigidity they are trapped in their conformation for kinetic ones [36].
A novel concept emerged from the above scenario. If we employed lyophilized protein powders and suspend them in organic solvents, would it be possible to encapsulate proteins within PLGA having a native secondary structure? To test this concept, we employed BSA and recombinant human growth hormone as model proteins. Figure 3 shows the outline of the experiments.
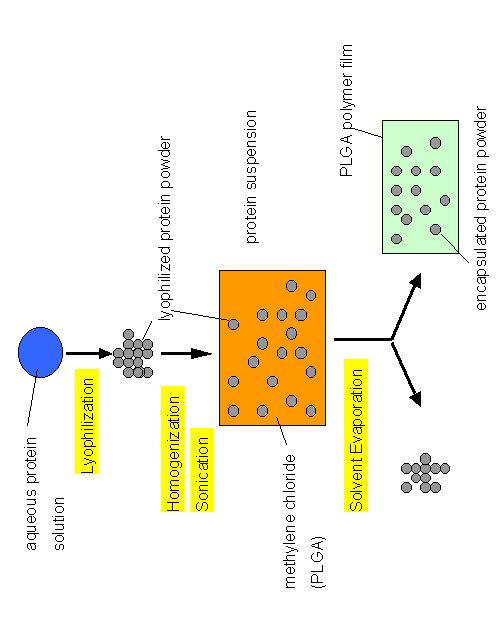
Figure 3. Encapsulation of dehydrated protein powders in PLGA
by a completly non-aqueous protocol.
It is important to note that the procedures we describe are not novel themselves. Encapsulation of protein powders in PLGA by simple solvent evaporation was actually one of the very first methods to be employed [17]. It leads to macroscopic delivery devices that are probably suitable for implanting purposes. However, due to their size, such devices are problematic because the creation of defined release profiles for the drug is very difficult. Nonetheless, the above simple experimental scheme demonstrated proof of concept because, in contrast to investigations using the w/o/w technique to encapsulate proteins in PLGA, it allowed us to investigate the protein secondary structure by FTIR spectroscopy at all stages in the encapsulation process. Thus, every process variable could be investigated separately and the detrimental effects on protein secondary structure minimized or completely circumvented.
Scenario 1: Employing protein in the absence of any stabilizer.
The first set of experiments was designed to analyze the fate of a protein lyophilized without any stabilizer using the above procedure. The protein secondary structure was determined both for the lyophilized powder, the powder is suspensed in methylene chloride and subsequently dried, and finally for the powder dispersed in a PLGA film. The latter was obtained by suspending the lyophilized protein in methylene chloride containing the dissolved polymer PLGA followed by direct casting onto a CaF2 FTIR window.
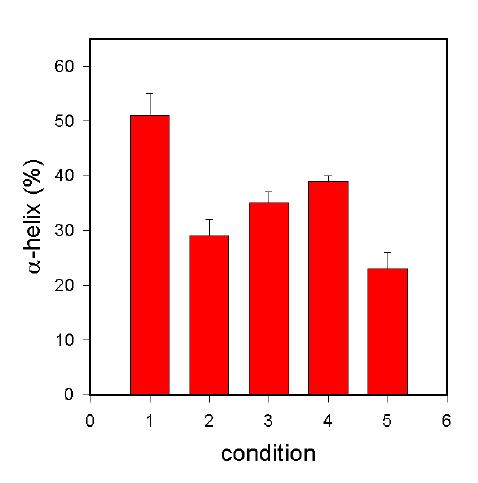
Figure 4. a-helix content of BSA at various stages in the non-aqueous encapsulation procedure.
For details see text below.
Figure 4 shows the fate of BSA when the protocol described above is carried out in the absence of any stabilizing additive. In aqueous solution, BSA has an a-helix content of ca. 57% (Bar 1). When BSA was lyophilized from such a solution, the a-helix content dropped significantly, in the case of our conditions to ca. 29% (Bar 2). If this protein was suspended by homogenization in methylene chloride and dried from this suspension, no further structural changes occurred, the a-helix content increased slightly (but statistically not significantly) to 35% (Bar 3). If this procedure was performed with PLGA dissolved in the solvent (and thus the final drying step caused encapsulation of BSA in the polymer matrix), the BSA structure was the same as it was without the polymer (39% a-helix, Bar 4). From this observation it can be concluded that the non-aqueous encapsulation procedure did not alter the structure of BSA significantly. The major structural changes occur during the lyophilization process. This is in contrast to the results obtained with the w/o/w procedure, where significant structural changes (in addition to those occurring upon lyophilization) are caused by the procedure. We also tested the impact of the method used to obtain the protein suspension, e.g. BSA was suspended by probe sonication in methylene chloride, whence the a-helix content was only 23% (Bar 5). Therefore, this method of creating the suspension of the protein in the organic solvent should be avoided.
From the above results one can surmise that, if lyophilization-induced structural changes are to be avoided, one should be able to obtain a protein with a more “native” secondary structure PLGA as a support.
Scenario 2: Employing protein in the presence of trehalose as a stabilizer.
Various lyoprotectants were tested for their ability to prevent lyophilization-induced structural changes in BSA. These results are available in our previous review (Griebenow et al., 1999). An additional difficulty encountered was that not all efficient lyoprotectants allowed the formation of a homogeneous suspension in the organic solvent. Trehalose proved to be the best additive in this context, even though structural preservation was superior employing sorbitol or glucose.
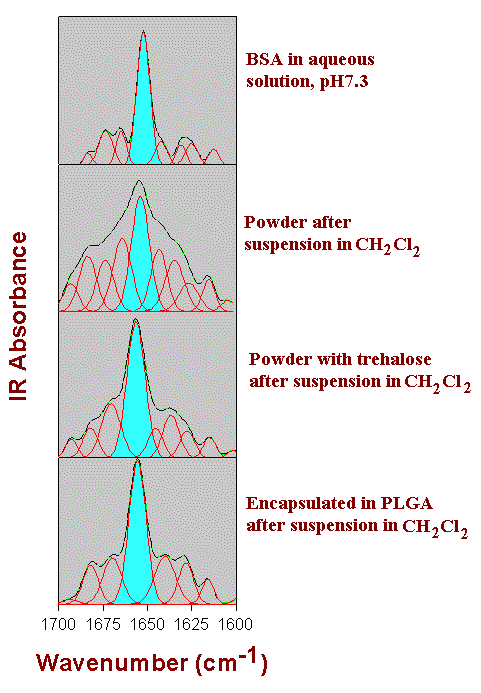
Figure 5. Amide I FTIR spectra of BSA after resolution-enhancement by Fourier-selfdeconvolution (black line), result of the Gaussian curve-fitting (green line), and individual Gaussian bands (red line). The a-helix band is shown in light-blue. For details, see text. (Figure modified after Carrasquillo et al., [27].)
Figure 5 shows the results of the FTIR investigations. At the top, the spectrum we show the amide I band region obtained for BSA in aqueous solution. As is typical for proteins with a high a-helix content, the spectrum is dominated by a band located at around 1655-1660 cm -1. When assigning this band to the a-helix secondary structure, the result of the spectral analysis by Gaussian curve-fitting is in excellent agreement with the X-ray structural data. The next two spectra show BSA lyophilized with and without trehalose after suspension in methylene chloride and subsequent drying. It is important to note that the spectra are extremly similar to those of the preparations formed directly after lyophilization. Therefore, the spectra are representative examples for both situations. For the preparation obtained in the absence of trehalose (the second spectrum), significant spectral changes in the amide I demonstrate severe structural changes. In particular, the amide I band and its component show broadening. However, when the protein is co-lyophilized with trehalose, the same protocol produced a dramatically different result. The a-helix band is again clearly the dominant band of the spectrum and band-broadening is much reduced compared to the former situation. In other words, the secondary structure of the protein was largely preserved. Finally, the ultimate spectrum shows BSA encapsulated in a PLGA polymer film. The spectrum is again very similar to that of the protein in aqueous solution. Therefore, we can conclude that, when the encapsulation procedure is optimized, the protein structure can be significantly improved upon encapsulation in the PLGA polymer matrix.
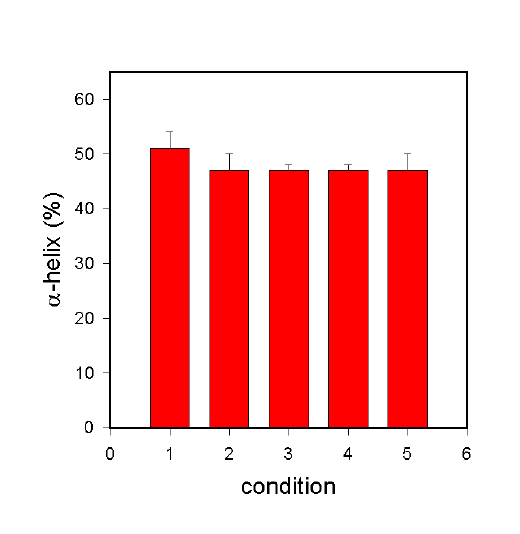
Figure 6. a-helix content of BSA at various stages in the non-aqueous encapsulation procedure. For details see text.
In the following, the section qualitative observations made above are supported by quantitative data (a-helix content). Figure 6 shows the fate of the BSA when the protocol in Fig. 3 is carried out in the presence of the stabilizing additive trehalose (data from Carrasquillo et al., [27]). The a-helix content of BSA in aqueous solution is shown in Bar 1 and is 51%. Lyophilization of BSA in the presence of trehalose at a 1:4 weight ration (BSA:trehalose) largely prevents lyophilization-induced structural changes and the a-helix content determined was indeed 47% (Bar 2). Suspension of this preparation by homogenization in methylene chloride and subsequent drying did not induce any additional structural changes. The a-helix content was remarkably still 47% (Bar 3). In addition, encapsulation of this formulation in PLGA using homogenization (Bar 4) or a sonication bath (Bar 5) did not produce any significant structural changes.
Conclusions
FTIR spectroscopy can be used to follow the fate of the model protein BSA in all the various encapsulation steps described. Combining the potential of novel encapsulation strategies with that of FTIR spectroscopy enable us to establish the structure-guided optimization of encapsulation conditions. Strategies have now been developed leading us to a significantly improved secondary structure of the protein in the PLGA polymer matrix. Similar investigations have also been performed with the model protein recombinant human growth hormone and lead us to identical conclusions [30].
Outlook
The technology we describe is currently being used to microencapsulate model proteins in PLGA. Herein, two immiscible organic solvents are used to create a single emulsion. The procedure is completly non-aqueous. BSA-loaded microspheres have already been obtained. Addional challenges have been encountered, e.g., the low yield of microspheres. In this context, the FTIR data collected previously have demonstrated the feasibility of our approach.
Acknowledgements
The authors acknowledge support by NIH-MBRS program (S06 GM08102-26S1), by the GAANN program of the US Department of Education, and the University of Puerto Rico (FIPI program).
References
- Thayer, A.M. (1995) Technology-based firms define new business approach to drug development. C&EN, June 5, 17-26
- Stinson, S. (1997) Growth factor protein may treat diabetes. C&EN, Nov. 24, 67-68
- Tamura, Y., Peng, P., Liu, K., Daou, M., and Srivastava, P.K. (1997) Immunotherapy of tumors with autologous tumor-derived heat shock protein preparations. Science, 278, 117-120
- Dickman, S. (1998) Cancer therapy. Antibodies stage comeback in cancer treatment. Science, 280, 1196-1197
- Robinson, J.R. (1997) Controlled drug delivery. Past, present, and future. In: Controlled Drug Delivery. Challenges and Strategies (K. Park, ed.), ACS Professional Reference Books, ACS, Washington DC, pp. 1-7
- Lee, J.J. (1995) Biopharmaceutical properties and pharmacokinetics of peptide and protein drugs. In: Peptide-Based Drug Design. Controlling Transport and Metabolism (M.D. Taylor and G.L. Amidon, eds.), ACS Professional Reference Book, ACS, Washington, DC, pp. 69-100
- Langer, R. (1990) New methods of drug delivery. Science, 249, 1527-1533
- Langer, R. (1993) Polymer-controlled drug delivery systems. Acc. Chem. Res. 26, 537-542
- Langer, R. (1995) Biomaterials and biomedical engineering. Chem. Eng. Sci. 50, 4109-4121
- Langer, R. (1995) 1994 Whitaker Lecture: Polymers for Drug Delivery and Tissue Engineering. Annals of Biomededical Engineering 23, 101-111
- Cleland, J.L. and Langer, R. (1995) Formulations and delivery of proteins and peptides: design and development strategies. In: Formulation and Delivery of Proteins and Peptides (J.L. Cleland and R. Langer, eds.), ACS Symposium Series, ACS Books, Washington DC
- O’Hagan, D.T., Rahman, D., McGee, J.P., Davis, H., and Williams, P. (1991) Biodegradable microparticles as controlled release antigen delivery systems. Immunology 73, 239-242
- Cohen, S., Alonso, M.J., and Langer, R. (1994) Novel approaches to controlled-release antigen delivery. Int. J. Technol. Assessment in Health Care 10, 121-130
- Saltzman, W.M. (1993) Antibodies for treating and preventing diseases: the potential role of polymeric controlled release. Crit. Rev. Therapeutic Drug Carrier Systems 10, 111-142
- Kissel, T., Li, Y.X., Görich, S., and Koneberg, R. (1996) Parenteral protein delivery systems using biodegradable polyesters of ABA block structure, containing hydrophobic poly(lactide-co-glycolide) A blocks and hydrophilic poly(ethylene oxide) B blocks. J. Controlled Release 39, 315-326
- Pearlman, R. and Nguyen, T. (1992) Pharmaceutics of protein drugs. J. Pharm. Pharmacol. 44 (suppl. 1), 178-185
- Langer, R. and Folkman, J. (1976) Polymers for sustained release of proteins and other macromolecules. Nature 263, 797-800
- Schwendeman, S.P., Cardamone, M., Brandon, M. R., Klibanov, A. M., and Langer, R. (1996) Stability of proteins and their delivery from biodegradable polymer microspheres. In: Microparticulate Systems for the Delivery of Proteins and Vaccines (Cohen, S. and Bernstein, H., eds.), Marcel Dekker, New York, pp 1-49
- Schwendeman, S.P., Costantino, H.R., Gupta, R.K., and Langer, R. (1997) Peptide, protein, and vaccine delivery from implantable polymeric systems. Progress and challenges. In: Controlled Drug Delivery. Challenges and Strategies (Park, K., ed.), American Chemical Society, Washington, D.C., pp. 229-267
- Arakawa, T., Prestrelski, S.J., Kenney, W.C., and Carpenter, J.F. (1993) Factors affecting short-term and long-term stability of proteins. Advanced Drug Delivery Reviews 10, 1-28
- Cleland, J.L., Lim, A., Barron, L., Desjardin, N., Duenas, E.T., Eastman, D.J., Vennari, J.C., Wrin, T., Berman, P., Murthy, K.K., and Powell, M.F. (1998) Development of a single shot subunit vaccine for HIV-1. 5. Programmable in vivo autoboost and long lasting neutralizing response. J. Pharm. Sci. 87, 1489-1495; Cleland, J.L. (1999) Single-administration vaccines: controlled-release technology to mimic repeated immunizations. Trends in Biotechnology 17, 25-29
- Schwendeman, S.P., Costantino, H.R., Gupta, R.K., Klibanov, A.M., and Langer, R. (1997) Peptide, protein, and vaccine delivery from implantable polymeric systems. In: Controlled Drug Delivery. Challenges and Strategies (K. Park, ed.), ACS Professional Reference Books, ACS, Washington DC, pp. 229-267
- Manning, M.C., Patel, K., and Borchardt, R.T. (1989) Stability of protein pharmaceuticals. Pharm. Res. 6, 903-918
- Volkin, D.B. and Klibanov, A.M. (1989) Minimizing protein inactivation. In: Protein Function: A Practical Approach (Creighton, Ed.). Oxford University Press, pp. 1-24
- Schöneich, C., Hageman, M.J., and Borchardt, R.T. (1997) Stability of peptides and proteins. In: Controlled Drug Delivery. Challenges and Strategies (K. Park, ed.), ACS Professional Reference Books, ACS, Washington DC, pp. 205-228
- Krishnamurthy, R. and Lumpkin, J.A. (1998) Stability of proteins during manufacture and release from biodegradable polymers. BioPharm. 11(1), 32-36
- Carrasquillo, K.G., Cordero, R.A., Ho, S., Franquiz, J.M., and Griebenow, K. (1998) Structure-guided encapsulation of bovine serum albumin in poly(DL-lactic-co-glycolic)acid. Pharm. Pharmacol. Commun. 4, 563-571
- Fu, K., Griebenow, K., Hsieh, L., Klibanov, A. M., Langer, R. (1999) Use of FTIR spectroscopy to characterize the secondary structure of proteins encapsulated within PLGA microspheres. J. Contr. Rel. 58, 357-366
- Costantino, H.R., Carrasquillo, K.G., Cordero, R.A., Mumenthaler,M., Hsu, C.C., and Griebenow, K. (1998) The effect of excipients on the structure and stability of lyophilized recombinant human growth hormone (rhGH). J. Pharm. Sci. 87, 1412-1420
- Carrasquillo, K.G., Costantino, H.R., Cordero, R.A., Hsu, C.C., and Griebenow, K. (1999) On the structural preservation of recombinant human growth hormone in a dried film of a synthetic biodegradable polymer. J. Pharm. Sci. 88, 166-173
- Yang, T.-H., Dong, A., Meyer, J., Johnson, O.L., Cleland, J., and Carpenter, J.F. (1999) Use of infrared spectroscopy to assess secondary structure of human growth hormone within biodegradable microspheres.
- Lapanje, S. (1978) Physicochemical aspects of protein denaturation. Wiley, New York.
- Privalov, P.L. (1992) Physical basis of the stability of the folded conformation of proteins. In: Protein Folding (Creighton, T.E., ed.), Freeman, New York, pp. 83-126
- Zaks, A. and Klibanov, A. M. (1984) Enzymatic catalysis in organic media at 100°C. Science 224, 1249-1251
- Volkin, D. B., Straubli, A., Langer, R., and Klibanov, A. M. (1991) Enzyme thermo-inactivation in anhydrous organic solvents. Biotechnol. Bioeng. 37, 843-853
- Griebenow, K. and Klibanov, A.M. (1996) On protein denaturation in aqueous-organic but not in pure organic solvents. J. Am. Chem. Soc. 118, 11695-11700
REF: K.Griebenow, I.Castellanos and K.G. Carrasquillo Int. J. Vib. Spect., [www.irdg.org/ijvs] 3, 5, 2 (1999)