Characterization of cerium dioxide dip-coated films by spectroscopic technique
CONTRIBUTED ARTICLE
6. Characterization of cerium dioxide dip-coated films by spectroscopic technique
Zorica Crnjak Orel
National Institute of Chemistry
Hajdrihova 19,
SI-1000 Ljubljana,
Slovenia
Email:zorica.crnjak.orel [at] ki.si
Introduction
Cerium oxide derived systems prepared by different methods as beam evaporation, reactive magnetron sputtering have been studied and used in various areas such as catalysis, optical additives, ionic conduction and for fuel cells [1-6]. As the rare-earth oxide [6] with high refractive indicies is prefered for multilayer optical devices. It possess high transparency in the visible and near-infrared region. The fundamental absorption edge is very sharp (characteristic of rare-earth oxide systems) with low luminous absorptance. It is also semiconductor with band gap Eg at ~3.2 eV. In recent years nanometer-sized particles have attracted much attention. Their chemical and physical properties are significantly differnt from bulk materials and have a great potential to become new materials useful for fine UV absorbent and high-activity catalysts [2]. Partial replacement of CeIV ions with LnIII ions in the lattice crates a corresponding number of anion vacancies and the resulting solid solution have high ionic conductivity [3]. In fuel cell doped ceria is a leading candidate for electrolyte [4].
Sol-gel deposited CeO2 and CeO2/SnO2 based films were also studied as counter electrodes in electrochromic devices [7-9] used for “smart windows”. The counter electrode provides electrochemical balance of the charge transferred during the oxidation and reduction reactions performed on primary electrochromic element. The inserted/extracted charge was twice as large for films containing 17 mol% SnO2 if compared to pure CeO2.
Due to the similar optical and electrical properties of CeO2 and TiO2 films, dye-sensitized CeO2 and mixed CeO2/SnO2 films using Ru (by)-complex were used as photoanodes in photoelectrochemical solar cells (new generation of nanophase solar cells) [10]. Cerium dioxide (CeO2) and CeO2/SnO2 films were employed as intercalation electrode. Stable and moderate cost solid-state galvanic cells with good stability was prepared with some solid polymeric electrolyte [11].
With the sol-gel method it is possible to prepare, at low temperature different materials as thin films or powders, through the formation of a colloidal suspension of metal oxies. The chemical homogeniety is maintained on an atomic scale for multicomponent systems. It is also one of the promising process for preparation of small particles with grain size in the nanoscale range.
Cerium dioxide (CeO2) crystallizes in the fluorite structure and consists of three sublattices [12]. The space group is Oh5. The primitive cell contains only one formula unit. This structure, with six optical-phonon branches, shows three zone-center frequencies: at 272 cm-1 (doubly degenerate TO mode), at 465 cm-1(triply degenerate Raman active mode) and at 595 cm-1 (nondegenerate LO mode) [13].
In this study the aqueous-based process was used for the preparation of pure CeO2 and mixed CeO2/SnO2 oxides. The method is inexpensive (matal salts already available) compared to the alcohol-based methods (mainly expensive metal alkoxide as starting materials) [14]. Prepared metal sols we used for the deposition as a thin films on indium tin oxide (ITO) plates (glass plates precoated with In2O3:Sn) for counter electrodes in electrochromic devices [7-9]. In the present work the influence of added SnO2 on the CeO2 thin films, previously investigated [14-16] for counter electrodes was determined by X-ray diffraction, FT-IR and Raman spectroscopies.
Experimental
CeO2 and mixed CeO2/SnO2 were prepared via the sol-gel route from aqueous solutions of inorganic precursors of the metal salts Ce(NH4)2(NO3)6 (99%, Fluka, Switzerland) and SnCl4 (7) (98 %, Alfa Product, Germany). Precipitates were obtained by the addition of NH4OH (pH=9) (Kemika, Croatia) and washed by bidistilled water in order to remove residual NH4+ and Cl– ions. The peptization was performed according to a modified literature method [17], by the addition of an equimolar quantity of HNO3 (pH » 1) (18) (Merck, Germany) and aging up to 90oC. Films were prepared by the dip-coating method on glass plates.
The FT-IR spectra at 4 cm-1 resolution were recorded on a Digilab FTS-80 spectrometer. Spectra of sols were obtained after the deposition of sols between CdTe plates. After the evaporation of water the spectra of xerogels were obtained. Films on CdTe plates were heated for 5 minutes to obtain spectra at 300oC. The Fourier-transform (FT) Raman spectra (at resolution 4 cm-1) were measured with a Perkin-Elmer 2000 spectrometer. The FT-Raman scattering was generated by 0.2-1.2 W of CW radiation at 1064 nm from a diode-pumped Nd:YAG laser and collected in a back-scattering geometry.
X-ray diffraction analysis (XRD) of the dip-coated films was performed on a Philips PW1710 X-ray diffractometer (X-ray source; CuKa ). The mean crystallite size was evaluated from X-ray diffraction peaks by the Scherrer equation [19].
Results and Discussion
Dip-coated films of pure CeO2 and mixed CeO2/SnO2 were prepared from non-aggregated sols using nitric acid for peptization because this gave dense oxide packing. It has been reported that the shape and size of colloidal particles [20] may be controlled by an addition of different anions. Sols prepared by the dispersion method are usually peptized with nitric acid. The rate of peptization depended upon the amount of acid added [21] A very important parameter in controlling the stability of a sol is the ratio of amounts of anion to metal oxide. The density of CeO2 [18]) was highest if the molar ratio of HNO3/CeO2 was one.
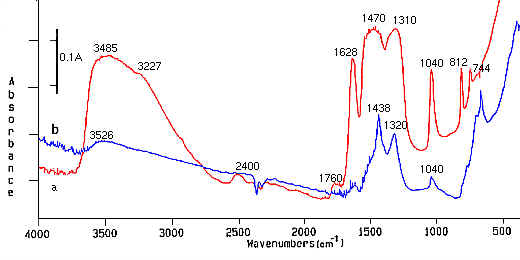
Figure 1. IR spectra of: a) gel/xerogel of cerium dioxide, b) cerium dioxide after heating at 300oC.
The influence of NO3 ions on our prepared films (CeO2 and CeO2/SnO2) was followed by FT-IR and Raman spectroscopy [22,23]. IR spectra of sol/xerogel and heated samples at 300oC of pure CeO2 after the addition of 17 mol% of SnO2 are presented in Figure 1. The wavenumber and width of the nitrate peak changed with the transformation of sol to gel/xerogel or heated samples. The bands at 1470 cm-1, 1310 cm-1 and 1040 cm-1 in Figure 1a for gel/xerrogel confirmed the bidentate complex formation of NO3– groups due to the separation (160 cm-1) of the two highest frequency bands at 1470 cm-1 and 1310 cm-1[24,25]. The band at 3410 cm-1 is attributed to the OH mode belonging to the hydrated species, and bands at 812 cm-1 and 744 cm-1 arose from the HNO3molecules [26]. The band at 1760 cm-1 also prove bidentate complex formation of NO3– groups in gel/xerogel. An ionic nitrate group at that frequency shows one relatively strong and sharp band, whilst a coordinated nitrate group contains two separate bands [27]. A decrease in intensities for NO3 coordination was reported as ionic>unidentate>bidentate [24,27].
These results have confirmed the bidentate complex formation of NO3– groups when the samples were prepared as gel/xerogel. It also appears that NO3–coordination in samples changes (as evidenced by shifts on the band) after the further heating of the samples. Band from 1470 cm-1 was shifted to 1438 cm-1 as presented in Figures. 1a and 1b, respectively. The separation of two highest frequency band from 160 cm-1 (Figure 1a) after heating for 5 minutes was only 118 cm-1, which can be understood as unidentate complex formation [24]. Very similar spectra were obtained for sol-gel/xerogel transformation of pure CeO2. After heat treatment at 300oC, the samples showed bidentate complex formation of NO3 groups.The influence of NO3 ions on our prepared films (CeO2 and CeO2/SnO2) was followed by FT-IR and Raman spectroscopy [22,23]. IR spectra of sol/xerogel and heated samples at 300oC of pure CeO2 after the addition of 17 mol% of SnO2 are presented in Figure 1. The wavenumber and width of the nitrate peak changed with the transformation of sol to gel/xerogel or heated samples. The bands at 1470 cm-1, 1310 cm-1 and 1040 cm-1 in Figure 1a for gel/xerrogel confirmed the bidentate complex formation of NO3– groups due to the separation (160 cm-1) of the two highest frequency bands at 1470 cm-1 and 1310 cm-1[24,25]. The band at 3410 cm-1 is attributed to the OH mode belonging to the hydrated species, and bands at 812 cm-1 and 744 cm-1 arose from the HNO3molecules [26]. The band at 1760 cm-1 also prove bidentate complex formation of NO3– groups in gel/xerogel. An ionic nitrate group at that frequency shows one relatively strong and sharp band, whilst a coordinated nitrate group contains two separate bands [27]. A decrease in intensities for NO3 coordination was reported as ionic>unidentate>bidentate [24,27].
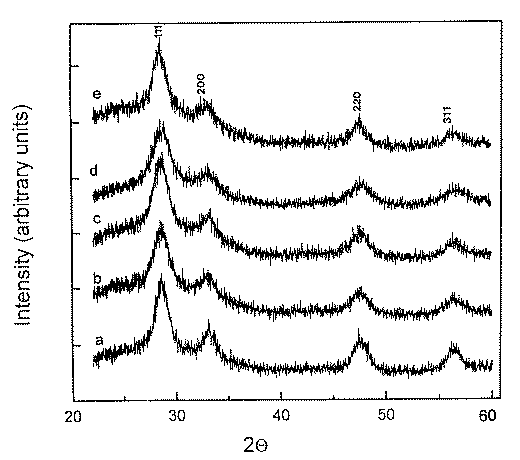
Figure 2. X-ray diffraction patterns of films deposited by dip-coating method and heat treated at 500oC: pure CeO2, b) CeO2after addition at 6.25 mol% of SnO2, c) CeO2 after addition at 9 mol% of SnO2, d) CeO2 after addition of 17 mol% of SnO2, e) CeO2 after addition of 50 mol% SnO2.
The bidentate complex formation of NO3 groups in gel/xerogel was confirmed by Raman spectroscopy (22). In the Raman spectra after heating the xerogels at 300oC the 1050 cm-1 band of NO3– groups is still present. We prepared our samples at 500oC. At that temperature in Raman spectra was no evidence of NO3– groups. At around 460 cm-1 was obtained T2g mode of the Raman active vibrations of CeO2 (22, 28). X-ray diffraction measurements of thin films heated at 500oC, the same as for preparations of crystalline cerianite with peaks at 2q = 28.6, 33.34, 47.5 and 56.5 (CuKa ). CeO2 and CeO2/SnO2 thin films prepared at that temperature possessed the best intercalation properties for Li+ ions [7]. From the results obtained by X-ray diffraction (Figure 2), the mean crystallite size (Scherrer relation) for about 300 nm thick films, calcined at 500oC, were calculated and are presented in Table I. Results obtained are comparable with the previous reported results of mean crystallite size for Ce(IV) precipitate [20]) and ceria gel [11] where mean crystallite size was 6.5 nm and did not change after the evaporation of some water (sol-gel conversion).
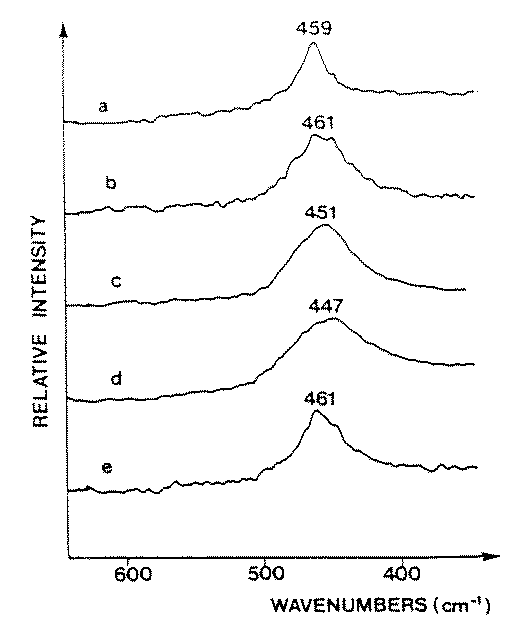
Figure 3. Raman spectra of films deposited of dip-coating method and heat treated at 500oC:a) pure CeO2, b) CeO2 after addition at 6.25 mol% of SnO2,
c) CeO2 after addition at 9 mol% of SnO2, d) CeO2 after addition of 17 mol% of SnO2, e) CeO2 after addition of 50 mol% SnO2.
We used the same dip-coated films also for an FT Raman spectroscopy study (Figure 3). In order to characterize exactly the same films as in X-ray the thin films were scraped from the glass substrate. A diamond screper was needed due to the very good adhesion and hardness of the films. The obtained results are presented in Table I. The Raman T2g mode shows some broadening and shifts to lower frequency when mean crystallite size calculated from X-ray difraction, decreased. No peaks f[29] found a strong correlation between the Raman linewidth and the inverse particle size for CeO2 particles in automotive exhaust-gas catalysts. They obtained an empirical relation for particle size down to 7 nm when half-width at half maximum of Raman linewidth was plotted as a function of the inverse of the particle size (Scherrer relation). Similar changes were observed for very small crystallites (2-20 nm) in CeO2/Al2O3 samples which were attributed to the smaller particle size. This phenomenon was explained by the relaxation of the momentum conservation selection rule for smaller particle size [30].
| Addition of SnO2 (mol%) |
Half-width from Raman spectra (cm-1) |
X-ray D Å |
| 0 | 20 | 70 |
| 6.25 | 36 | 62 |
| 9.0 | 50 | 60 |
| 17 | 60 | 45 |
| 50 | 31 | 66 |
Table 1. Raman and X-ray data of cerium dioxide (CeO2) and mixed CeO2/SnO2.
From obtained results we confimed that the CeO2 particles size after the addition of SnO2 can be qualitatively determined by Raman spectroscopy. If we compare the time needed for obtained X-ray measurements or transmission electron microscopy (TEM) data which are usually applied to particle size determination, the corresponding Raman spectra can be obtained much quicker.
Conclusion
The bidentate complex formation of NO3– ions in gel/xerogel CeO2 and CeO2/SnO2 films was confirmed. After the heating at 300oC in CeO2/SnO2 it change to unidentate complex formation.
Changes in primary particle size in CeO2 and mixed CeO2/SnO2 films, prepared via the sol-gel route were determined by X-ray diffraction and qualitatively correlated with FT-Raman spectroscopy. The mean crystallite size of pure CeO2and CeO2 mixed SnO2 films was calculated by the Scherrer relation. A strong correlation between the inverse of the particle size and Raman linewidth has been confirmed. As the particle size decreases the Raman allowed line shifts and broadened.
Acknowledgement
This work was supported by the Ministry of Science and Technology, Republic of Slovenia (Research contract J2-1468).
References
- S. Imamura, Y. Uematsu, and K. Utani, T. Ito, Ind. Eng. Chem. Res. 30, 18 (1991).
- T. Masui, K. Fujiwara, K. Machida, and G. Adachi, T. Sakata, H. Mori, Chem.Mater. 9, 2197 (1997).
- A.K. Bhattacharya, A. Hartridge, K.K. Mallick, J.L. Woodhead, J. Mater. Sci. 31, 5005 (1996).
- A. Atkinson, Solid State Ionics 95, 249 (1997).
- T. Inoue, Y. Yamamoto, M. Satoh, A. Ide, S. Katsumata, Thin Solid Films281-282, 24 (1996).
- S.-Y. Zheng, A.M. Andersson-Fäldt, B. Stjerna, C.G. Granqvist, Appl. Optics 32, 6303 (1993).
- Z. Crnjak Orel and B. Orel, J. Mater. Sci., 30, 2284 (1995).
- P. Baunry, A.C.M. Rodrigues, M.A. Aegerter, and L.O. Bulhoes, J. Non-Cryst. Solids, 121, 319 (1990).
- R. David Rauh and S.F. Cogan, Solid State Ionics, 28-30, 1707 (1988).
- A. Turkovic, Z. Crnjak Orel, Solid State Ionics, 89, 255 (1996).
- A. Turkovic, Z. Crnjak Orel, Solar Energy Materials and Solar Cells, 45, 275 (1997).
- S. Michizuki, Phys. Stat. Sol. (b), 114, 189 (1982).
- W.H. Weber, K.C. Hass, and J.R. McBride, Phys. Rev. B, 48, 178 (1993).
- Z. Crnjak Orel, B. Orel, M. Hodošcek, and V. Kavcic, J. Mater. Sci., 27, 313 (1992).
- Z. Crnjak Orel, B. Orel, and M. Klanjšek Gunde, Solar Energy Materials, 26, 105 (1992).
- Z. Crnjak Orel, I. Muševic and B. Orel, Proc. “Nanoparticles in Solid and Solutions”, J.H. Fendler and I. Dékány (eds.), Kluwer Academic Publishers, 1996, pp. 519-528.
- A. Atkinson and R.M. Guppy, J. Mater. Sci., 26, 3869 (1991).
- R.L. Nelson, J.D.F. Ramsay, and J.L. Woodhead, J. A. Caoirns, J.A. Crossley, Thin Solid Films, 81, 329 (1981).
- F. Czerwinski and W.W. Smeltzer, J. Electrochem. Sci., 140, 2606 (1993).
- W.P. Hsu, L. Ronnquist, and E. Matijevic, Langmuir, 4, 31 (1988).
- C.W. Turner, Ceramic Bulletin, 70, 1487 (1991).
- Z. Crnjak Orel and B. Orel, Solar Energy Materials, 40, 205 (1996).
- Z. Crnjak Orel, Applied Spectrosc. 53, 241 (1999).
- A.A. Davydov, Infrared Spectroscopy of Adsorbed Species on the Surface of Transition Metal Oxides (John Wiley&Sons, Chichester, 1990).
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds (John Wiley&Sons, New York, 1978).
- D.R. Lombardi, C. Wang, B. Sun, A.W. Fountain III, T.J. Vickers, C.K. Mann, F.R. Reich, J.G. Douglas, B.A. Crawford, and F.L. Kohlasch, Applied Spectroscopy 48, 875 (1994).
- A.B.P. Lever, E. Mantovani, and B.S. Ramaswamy, Can. J. Chem. 49, 1957 (1971).
- V.G. Keramidas and W.B. White, J. Chem. Phys. 59, 1561 (1973).
- G.W. Graham, H.W. Weber, C.R. Peters, and R. Usmen, J.of Catalysis, 130, 310 (1991).
- J.Z. Shyu, W.H. Weber, and H.S. Gandhi, J. Phys. Chem., 92, 4964 (1988).
Received 19th August 1999, received in revised format 17th September,
accepted 20th September 1999.
REF: Orel Z., Int. J. Vib. Spect., [www.irdg.org/ijvs] 3, 4, 6 (1999)