Vibrational Microscopy and Imaging of Heterogeneous Inorganic Materials
5. Vibrational Microscopy and Imaging of Heterogeneous Inorganic Materials
Jon R. Schoonover,a George J. Havrilla,a Patrick J. Treadob
a) Los Alamos National Laboratory
Mail Stop J586
Los Alamos, NM 87544
USA
b) ChemIcon, Inc.
7301 Penn Avenue
Pittsburgh, PA 15208
USA
Abstract
Raman and infrared microscopy and imaging have been utilized to interrogate highly heterogeneous inorganic systems. The vibrational data provide structurally specific data that are useful in our understandinf of the underlying chemistry of these systems. One unique approach has been to couple vibrational microscopy and imaging with elemental images to gain additional insight into these samples.
Introduction
Raman spectroscopy uses laser light scattering to measure the vibrational frequencies of molecules, while infrared spectroscopy involves the absorption of infrared radiation to provide vibrational information. These measurements provide a spectroscopic “fingerprint” that reveals the identity and condition (oxidation state, molecular structure and speciation information) of molecules which constitute a material. Additionally, Raman spectroscopy also describes molecular conformational information (crystalline phase, degree of order, strain, grain size). The recent development of approaches to perform vibrational imaging, which combines the molecular analysis capabilities of Raman or IR spectroscopy with the visualization power of optical microscopy, provides a powerful method for obtaining molecular information in inorganic materials, especially highly heterogeneous materials.
Vibrational imaging experiments based on these spectroscopic techniques utilize either direct imaging or mapping. Direct imaging is accomplished by illuminating a large field of view of the sample and imaging the scattered (Raman) or transmitted (IR) light onto an imaging two-dimensional array detector [1,2] (e.g. a CCD camera; InSb or MCT focal plane array) positioned at the focal plane of the optical system. The mapping experiment involves measuring a spectrum at each point of a grid for well-defined spatial locations of a sample. An image can then be reconstructed from the spectrum at each location. In some instances, direct imaging and mapping are combined into hybrid methods as in line scanning imaging Raman spectrometers.
One area of research where vibrational microscopy and imaging can play a vital role is the disposition and treatment of various nuclear industry waste forms and research samples contaminated with plutonium. This type of sample typically consists of a highly heterogeneous distribution of inorganic species. Furthermore, because of the hazards associated with this type of sample, keeping a small sample size mitigates numerous safety concerns. One novel approach we have taken in these studies is to utilize elemental imaging approaches to direct and assist the vibrational microscopy and imaging studies.
The study of interactions and structural characteristics of plutonium complexes in different substrates is vital information in exploring stabilization, processing, and storage scenarios. The interactions of actinides with various waste forms and media, and the fate and transport of actinides, are areas of particular interest. In this paper, we demonstrate and discuss the use vibrational microscopy and imaging technology in (1) the study of plutonium in brine solution,
(2) characterizing an incinerator ash sample containing plutonium, and
(3) characterizing a mixed-oxide (MOX) plutonium surrogate sample.
Experimental
Raman microscopy, point-by-point imaging, and line scan imaging.
The basic experimental arrangement for Raman microscopy and point-by-point or line scan imaging utilize laser excitation at 488.0 or 514.5 nm supplied by a Spectra Physics 2025 Ar+ laser or 752 nm radiation from a Ti: Sapphire laser pumped by the 2025 Ar+ laser. This laser excitation is coupled to an infinity-corrected optical microscope (Zeiss Axiovert 135) using a holographic SuperNotch Plus rejection filter (Kaiser Optical Systems, Inc.). In the line scanning experiment a mirror in the optical path of the laser is operated in an oscillating mode to produce an excitation line. The microscope objective (Zeiss LD-EPIPL 20x/0.4 or Ziess PLAN-NEO 63x/0.95) is used to both focus the laser light and collect the Raman scattering. The collected light is passed back through the holographic notch filter and dispersed by a HoloSpec f/2.2 monochromator (Kaiser Optical Systems, Inc.) equipped with the appropriate holographic Raman grating and a liquid-nitrogen-cooled Photometrics PN12 CCD.
IR microscopy and imaging.
The FTIR microscopy and imaging (point-by-point mapping) have used a Spectra-Tech Research IRplan microscope, coupled to a Nicolet 20SXB FTIR bench, and equipped with a programmable Cell Robotics microscope stage. Synchronization of stage movement, data collection, and certain data processing steps are accomplished through the Nicolet MacrosPro software interface with samples examined in the transmission mode.
Direct Raman imaging.
Direct Raman imaging experiments were conducted using the Falcon Raman imaging microscope system (ChemIcon, Inc.). The Falcon system uses a diode pumped Nd:YVO4 solid state laser source doubled to operate at 532 nm (Spectra Physics, Millennia II) coupled with a multimode fiber optic relay to an infinity-corrected optical microscope (Olympus, BX60) via an illuminator assembly (ChemIcon, Raman Illuminator). The illuminator defocuses the laser source and illuminates the entire sample field of view through a 50X objective (Olympus, 0.80 N.A.). The Raman scattering is collected with the same objective and is transmitted back through the illuminator which houses holographic notch rejection filters to remove the Rayleigh scattering. The Raman signal is filtered with a 9-cm-1 bandpass liquid crystal tunable filter (LCTF) constructed using the Evans Split-Element geometry (CRI, VariSpec). Raman images are collected using a themoelectrically (Peltier) cooled (-40 oC) slow-scan charge-coupled device (CCD) detector (Princeton Instruments, TE/CCD-512TKB) that has 512 x 512 (20 mm square) pixels.
Elemental imaging.
Elemental images were measured using X-ray fluorescence spectrometry with a Kevex Omicron spectrometer equipped with a 50W rhodium X-ray anode oriented at a 45-degree angle with respect to the sample stage. The detector (a liquid nitrogen cooled lithium drifted silicon chip with an active area of 50 mm2) is also oriented at a 45-degree angle. Scanning a selected number of frames, and then co-adding the intensity from each frame in a dynamic mode can produce an elemental image. Elemental mapping analysis was also performed with a scanning electron microscope (SEM; RJ Lee Instruments) outfitted with secondary electron and backscatter electron detection capability. The elemental imaging was accomplished with the SEM using energy dispersive spectrometer with point analysis and elemental mapping capability.
Results & Discussion
Disposition of plutonium in brine solution.
Interactions of actinides with various media and their mobility in the media are areas of research where vibrational microscopy and imaging can play an important role. As an example, a project has been designed to study the behavior of actinides, particularly Pu, in brine. In this project, test containers with actinide contaminated waste are exposed to brine solutions (and a variety of chemical variables) to study the behavior of actinides in this medium. These test containers are periodically tested for soluble actinides and precipitated material. The containers were kept at constant temperature, rotated weekly for 15 min., and allowed to settle for 2 – 3 days. Brine solutions were extracted at selected intervals from the test container and filtered in series through 5-mm, 1-mm, and < 20-nm filters.
These filter samples represent a highly heterogeneous sample type where characterizing the molecular species present can assist in beginning to understand the chemistry of actinides in brine. Elemental imaging of the filters was performed to correlate elemental composition, and to identify regions of high actinide concentration. Raman and infrared measurements were then made on spatially distinct regions of the sample.[3]
As an example, one specific test container was designed to test the effect of CO2 pressure on high concentrations of actinides in brine. This test container consisted of actual contaminated waste and CO2 headspace at 870 psig (60 Bar). The precipitate on the filter paper was a grey powder with some darker particles. The analytical data (measured by X-ray fluorescence spectrometry) for several sets of precipitates measured over a one year period indicated between 20 and 60 mg/cm2 of Pu. High levels of Ca and Mg, moderate levels of Sr and Cl, and low concentrations of Fe and S were also observed.
The elemental image of the filter paper (Figure 1) demonstrates the pattern of Pu concentration. The structured form of the precipitate on the filter paper is due to the filter holder used in this high-pressure test. One interesting observation from the elemental data is the correlation between Pu and Sr.
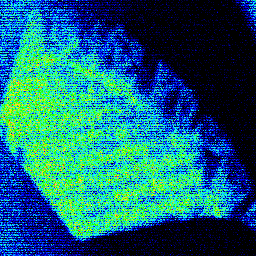
Figure 1. Micro-X-ray fluorescence image of plutonium distribution on a filter paper used to identify areas of high Pu concentration. The image is plotted on a thermal scale where the hotter colors represent higher Pu concentrations.
A representative micro-Raman spectrum (Figure 2, spot size ca. 1 mm) of an individual particle from the area of high Pu concentration shows a strong band at 1079 cm-1 with weaker features at 629, 698, 732, 1465, and 1537 cm-1. A second micro-Raman spectrum in this region shows bands at 732 and 1368 cm-1. The pattern of bands at 629, 698, 1078, 1465, and 1537 cm-1 is indicative of an inorganic carbonate as the dominant molecular species. This interpretation is confirmed with the micro-FTIR data (Figure 3, 50-mm spot size), which shows a strong feature centered near 1470 cm-1 and a sharp 876-cm-1 absorbance indicative of an inorganic carbonate. The IR data also demonstrates the presence of a hydrated inorganic sulfate with bands near 1150, 1620, and 3500 cm-1. Vibrational data coupled with the analytical and elemental imaging data indicate SrCO3 and CaSO4 . H2O are the primary inorganic complexes closely associated with areas of high Pu levels in the precipitate. The ability to identify these inorganic components in this highly heterogeneous waste forms assists in beginning to understand the complex chemistry in brine solutions under a CO2 atmosphere.
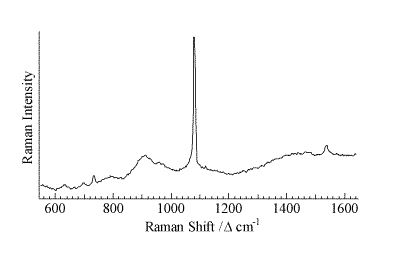
Figure 2.Micro-Raman spectrum of an individual particle of STTP precipitate in the area of high Pu concentration.
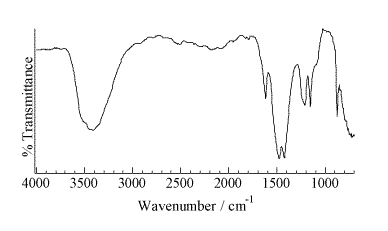
Figure 3.Micro-FTIR spectrum of a group of particles from STTP precipitate in the area of high Pu concentration
Characterization of an incinerator ash sample. An incinerator ash sample is another example of a Pu-contaminated material where understanding the molecular composition can lead to a better understanding of the chemistry. The goal is to identify the Pu species present and their relationship to other elements or molecular components in the sample. The sample of incinerator ash of actual plutonium contaminated waste is a fine gray-brown powder containing black particles.
Elemental maps of Pu concentration for a few particles of the ash sample demonstrate the distribution of Pu in this sample. The micro-Raman and -IR spectra were measured from areas of high Pu concentration and provide information of the Pu species and its relation to other inorganic complexes.[4] The most intense feature in the Raman spectrum is at about 475 cm-1. Actinide oxides are known to crystallize at high temperature with a fluorite (CaF2) structure and space group Fm3m(O5h) which possesses one Raman active phonon of T2g symmetry (478 cm-1 for PuO2) at k = 0. The 475-cm-1 Raman band can be assigned to this T2g mode.[5,6] The Raman data also suggest the presence of other oxide species such as SiO2, TiO2, and Al2O3. The micro-IR spectrum demonstrated by a strong band centered near 1050 cm-1 indicative of silica or a silicate as another major component of the ash sample.
In Figure 4, a Raman image from the 475-cm-1 Raman band is shown from an area demonstrating high Pu concentration from the elemental image of Pu. An overlay of the vibrational and elemental images provides a definitive assignment for the PuO2 as the predominant Pu species in the ash sample. A direct Raman spectrum collected through the LCTF from one of the micrometer-sized particles is also shown in Figure 4. The Raman and IR microscopy data of particles of high PuO2 concentration further demonstrates that SiO2 and PuO2 are closely associated in this sample with a heterogeneous distribution of additional metal oxide species. As with the precipitated material, the elemental images serve to direct the analysis to the area of interest in the sample. The molecular microscopy and imaging then supply further information on these spatially distinct regions. The data are interpreted as indicating that PuO2 is the dominant plutonium phase in this sample and is closely associated with silicate matrices such as SiO2.[7]
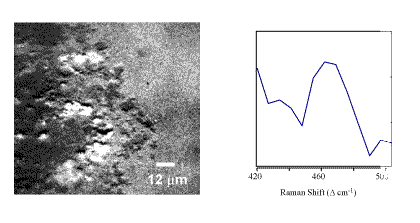
Figure 4.Ratioed Raman image (469/442 cm-1) of Pu(O2) in an incinerator ash sample along with the LCTF Raman spectrum of a micrometer-sized particle within the sample
Characterizing a residual Ga phase in a mixed-oxide (MOX) surrogate sample.
Combining scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM/EDS) elemental mapping and micro-Raman imaging represents an experimental approach applicable to identifying components distributed on the micron to submicron scale.[8] In this application, we demonstrate an initial integration of these techniques to examine and identify a residual gallium phase in a section of surrogate mixed oxide fuel feed pellet that had been sintered to remove the gallium.
Because of the proliferation issues associated with weapons-grade plutonium from dismantled weapons, conversion to a non-weapons grade material is highly desirable. Mixed oxide (MOX) fuel is one strategy available for using this surplus plutonium. MOX fuel production consists of converting plutonium metal to PuO2, which is then mixed with depleted UO2 and pressed into a fuel pellet to be burned in a light water reactor.
In the feed powders, it is also very important to minimize impurities; the most important ones are those that can interact with the fuel rod claddling and those that can absorb neutrons thereby limiting the performance of the MOX pellets. The use of weapons- grade plutonium in mixed oxide fuels is, therefore, dependent upon removal of any gallium. Gallium is added as an alloying agent in weapons-grade material, while in a MOX material it can be a corrosive agent via interactions with the fuel cladding material. To remove the gallium, the material is reduced by heating in the presence of H2 to generate Ga2O. This suboxide is volatilized above 600 oC and liberated as a vapour.
Research efforts have focussed on using a surrogate material consisting of CeO2 to mimic PuO2 spiked with Ga2O3to study the removal of gallium. CeO2 exhibits similar chemical and physical properties as PuO2. Initial studies using a thermal reduction process have shown that unfortunately a residual amount of gallium remains following this treatment. These studies demonstrated that the gallium aggregates along grain boundaries of the matrix material and the exterior of the surrogate pellet. Identifying the residual gallium phase is an important aspect of the overall study of MOX fuel fabrication.
Figure 5 compares the SEM/EDS elemental mapping of O, Ga, and Ce with the backscatter electron image at 550x magnification of a cross-sectioned surface of a surrogate MOX fuel feed pellet sintered at 1200oC. This mapping demonstrates oxygen distributed throughout the cross section, while gallium is localized in the grain boundary structures. Cerium is the major component of the grains, but is also present at lower levels in the grain boundaries. SEM images further indicate that a glassy amorphous phase is present in the grain boundaries. Ga, Ce, and O in the grain boundaries suggest some type of perovskite phase.

Figure 5.SEM/EDS elemental chemical imaging of sintered MOX surrogate pellet showing the elemental distribution of O (upper left), Ga (upper right), Ce (lower left), and a backscatter electron image (lower right)
Imaging Raman features of the MOX fuel feed pellet can provide further contrast between phases as well as information on the nature of the phases. Raman images using bands associated with Ga from the SEM/EDS data (292 cm-1, Figure 6) and CeO2 (464 cm-1, Figure 6) demonstrate that the 292-cm-1 intensity is indeed localized in the grain boundaries. The Raman images were processed using cosine correlation analysis (CCA). CCA is a multivariate technique that can reveal subtle spectral differences that are not immediately obvious in the raw images.[9] The CCA images provide contrast enhancement that clearly show a gallium phase in a CeO2 matrix.

Figure 6.Raman chemical images at 292 cm-1 (left) highlighting the distribution of the Ga phase, and at 464 cm-1 (right) indicating the distribution of CeO2.
A major advantage of LCTF-based imaging is the ability to produce Raman spectra from small regions of interest in the Raman images (Figure 7). The spectra demonstrate the characteristic F1g Raman band at 464 cm-1 for CeO2within the grains. A weaker series of bands including the 292-cm-1 feature are associated with the phase containing Ga.
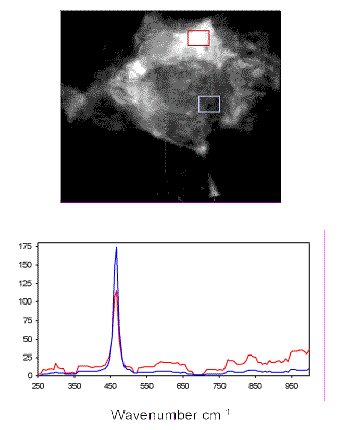
Figure 7. LCTF Raman spectra of MOX surrogate feed material from the indicated areas of the Raman image at 292 cm-1.
This data suggests a perovskite phase concentrated in the grain boundaries of the surrogate material. In the presence of CeO2, a perovskite structure is possible and could well be similar to other lanthanide aluminates and gallates (e.g. LaAlO3 and NdGaO3). The perovskite structure consists of oxygen ions and the larger lanthanide cations forming a cubic close packed array with the smaller cation (Al or Ga) occupying the octahedral holes formed by the O ions. This structure consists of the lanthanide cations occupying the corners of the cube, the oxides on the face of the cubes, and the small cation filling the center.
The Perovskite structures are often slightly distorted. If CeGaO3 is formed in the sintering process, this material can take on a cubic structure or a deviation from this structure. A cubic form similar to CeAlO3, for example, would give rise to lattice modes described as 4F1u + F2u. The acoustic mode is F1u, so the four vibrational modes are of symmetry 3F1u + F2u. The F1u modes are IR active, while the F2u mode is silent. There are no Raman active vibrational modes, so this structure or a slight distortion from this structure is expected to result in very weak or absent Raman bands. In the case of CeO2 on alumina reacted in a reducing environment at 920oC, the Raman spectrum demonstrates a band near 460 cm-1 superimposed on a broad fluorescence background.[10] Since cubic CeAlO3 gives no Raman active vibrations, the spectrum was interpreted as resulting from a mixture of CeO2 and CeAlO3.
The LCTF Raman spectrum of the Ga phase supports the idea that the Ga is present as a perovskite phase. The LCTF Raman spectrum is very different from the Ga2O3 starting material and literature spectra of a-Ga2O3 and b-Ga2O3. The LCTF Raman features are very weak and not observed without the aid of CCA. The combination of SEM and Raman imaging data is consistent with a perovskite structure in a cubic or nearly cubic geometry.
References
- B.W. Cook, Internet.J.Vib.Spect [www.irdg.org/ijvs] 2, 4, 2 (1998)
- Christopher T. Zugates and Patrick J. Treado, Internet.J.Vib.Spect [www.irdg.org/ijvs] 2, 4, 7 (1998)
- J. R. Schoonover, F. Weesner, G. J. Havrilla, M. Sparrow, and P. J. Treado, Appl. Spectrosc. 1998, 52, 1505 .
- J. R. Schoonover and G. J. Havrilla, Appl. Spectrosc. 1999, 53, 257.
- G.M. Begun, R.G. Haire, W.R. Wilmarth, and J. R. Peterson, J. Less-Common Met. 1990, 162, 129
- V.G. Keramidas and W.B. White, J. Chem. Phys. 1973, 59, 1561.
- R.G. Behrens, E.C. Buck, N.L. Dietz, J.K. Bates, E. Van Deventer, and D.J. Chaiko, Argonne National Laboratory Report ANL-95/35, 1995.
- C. T. Zugates, A. S. Bangalore, and P. J. Treado, “Raman/SEM Chemical Imaging of Heterogeneous Inorganic Materials,” ChemIcon Report 1998.
- H. R. Morris, J. F. Turner II, B. Munro, R. A. Ryntz, and P. J. Treado, Langmuir 1999, 15, 2961.
- J.Z. Shyu, W.H. Weber, and H.S. Gandhi, J.Phys. Chem. 1988, 92, 4964.
Received in original format 28th May 1999,accepted 7th June 1999.