Progress in instrumentation for Raman micro-spectroscopy and Raman imaging for cellular biophysics

8. Progress in Instrumentation for Raman micro-spectroscopy and Raman imaging for cellular Biophysics
C. Otto and J. Greve
University of Twente,
Department of Applied Physics,
Biophysical Technology group,
Institute for BioMedical Technology,
P.O.Box 217,
7500 AE Enschede,
The Netherlands
Abstract
The progress in instrumentation for Raman micro-spectroscopy and Raman imaging has reached standards that allow application of these methods in material sciences ranging from the bulk of solid state, liquid and gaseous phases towards more subtle and complex systems like interfaces, reacting chemicals, biological cells and tissues. The purpose of this paper is to describe recent instrumental developments in Raman microscopic instrumentation. As an illustration spectra and images of a chromosome will be presented.
Introduction
The combination of microscopy with Raman light scattering, as proposed early in the seventies [1], is a key-idea in the biological applications of Raman spectroscopy. With this approach it is possible to obtain Raman spectra of minute amounts of material from factually any location that can be accessed by light. Initially two problems remained that halted the progress in cell biological applications. The first problem was the fluorescence problem and the second problem was the background of Raman scattered light generated in “other-locations-than-the-desired-location” from for instance immersion fluids or substrate. A different problem, intimately involved with the application of laser techniques in cellular biology, is the desire to keep cells alive and intact, at least during the measurement time. Raman micro-spectroscopy often allows such non-invasive measurements [2]. Solutions are found in a proper choice of experimental conditions, based on the knowledge of the biological specimen, its optical characteristics and the information that is desired.
The first problem has been solved by the use of red laser light sources and extremely sensitive CCD-cameras capable of measuring, by photon counting, many scattered frequencies simultaneously. The use of red laser light sources is not a stringent requirement when the desired information can be acquired in a sufficiently short time. The second problem has been solved by the implementation of the confocal measurement principle in Raman micro-spectroscopy [3]. The maximal resolution (or minimal observable distance between two object points) in the lateral (x,y) direction and in the axial (z) direction can be calculated from the following (approximate) formulas (see for a thorough discussion [4]):
![]()
Using laser light with a wavelength l =647.1 nm and a water (n=1.33) immersion objective with a numerical aperture NA=1.2 the lateral resolution becomes 329 nm and the axial resolution becomes 1195 nm. In real life biological samples two separate objects rarely occur and one deals with a continuous and in-homogeneous scattering medium, typically a living cell. To achieve said resolutions the confocal measurement principle is introduced (Figure 1). The light scattered from the object in plane “0” is transmitted through the pinhole most efficiently. Light scattered from planes above (plane A) and below (plane B) is respectively focused beyond and before the pinhole, resulting in an attenuation of the scattering from these planes behind the pinhole. For weak signals, typical in Raman light scattering, it is extremely important that only desired signal photons reach the detector. This is accomplished to some degree by measuring confocal.
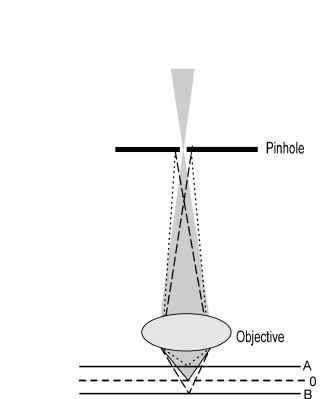
Figure 1. A pinhole in the image plane of the objective introduces the confocal principle. Light is selected from a thin layer improving the
signal-to-noise ratio of signal from the “confocal” section around plane “O”.
The use of two-dimensional detectors, such as a CCD camera, allows economic data collection. In principal Raman spectroscopy can be viewed as a six dimensional type of spectroscopy, much the same as fluorescence spectroscopy. Three dimensions for spatial coordinates. Two spectral dimensions, resp. one for the frequency of the exciting laser light and one for the frequency of the scattered light. The sixth dimension is time. The present two-dimensional detectors employed in Raman spectroscopy are used to measure simultaneously: (A) one spatial dimension and one spectral dimension resulting in the method of Raman line scanning and (B) two spatial dimensions resulting in the method of Raman direct imaging. The next spatial dimension is introduced by moving the object through the confocal region. The dimension time is added by measuring consecutive data-arrays.
Raman instrumentation was previously developed that was capable to record Raman spectra from a small focal region (< 10-15 L) inside cells using red (~660 nm) laser light [2,3]. Recently a Raman micro-spectrometer was developed that was also capable to record resonance Raman spectra excited with blue (413.1 nm) laser light [5,6] from single cells. The combination of living cells and excitation wavelengths in the green – down to the violet spectral region is not straightforward as living cells may quickly change their behaviour and sometimes even disintegrate. The resonance Raman effect results in a much higher signal flux. This makes data accumulation possible in a much shorter time. Enzymatic reactions in single, living cells could thus be observed using measurement times of 10 seconds/cell [5,6].
We have now developed two set ups: one for Raman micro-spectroscopy in Raman line scanning mode [7] and one for Raman micro-spectroscopy in Raman direct imaging mode [8]. Application has been to (bio-)materials for implantation technology [7,9], chromosomes [7,8], drugs in cells [8,10,11] and cataractous spots in eye-lenses [10,12].
New Raman instruments and results
The Raman line scanning spectrometer is schematically presented in Figure 2. The microscope is represented by an objective only. A line is imaged onto the sample by a scanning mirror driven by a piezo-electric transducer. The mirror orientation is modulated with approximately 40 Hz. Using a water immersion objective (Zeiss Neofluar, NA=1.2), with a magnification of 63 times, a line on the object is generated of 30 m m length. The back-scattered light, collected by the objective, passes through a slit. After the slit the beam of light is made parallel. Spectral filtering suppresses the back-scattered laser light. The grating is properly filled after beam expansion and the various frequencies in the scattered light are dispersed in one direction of the camera. In the perpendicular direction the spatial information is present. The use of a slit instead of a pinhole results in a slightly lower resolution [13], particularly in the axial direction.
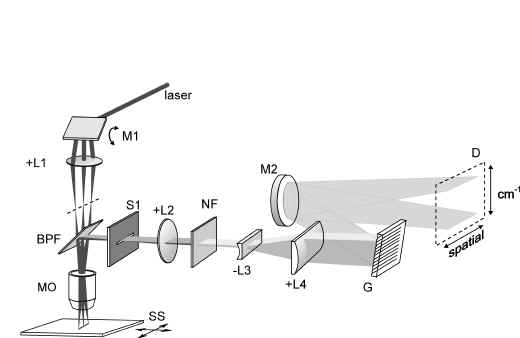
Figure 2. The Raman line scanning spectrometer. M1: scanning mirror, +L1: positive lens, BPF: Band pass filter, MO: microscope objective, S1: slit, +L2: positive lens,
NF: notch filter, -L3: negative lens, +L4: positive lens, G: grating, M2: mirror,
D: detector (Wright Instruments back-thinned LN/TBK512).
In Figure 3A. a Raman line scan image is presented of a part of a giant chromosome of Chironomus Thummi Thummi. The vertical direction coincides with spatial information (scale in m m) and the horizontal direction with spectral information. In Figure 3B. the white light image of the same chromosome is presented. The dark bands coincide with more condensed regions of the chromosome. The interbands are regions with higher transcriptional activity. Bright vertical lines in Figure 3A. are measured at frequencies with intense Raman scattering. The band at 1094 cm-1 is due to the phosphate stretching motion in the direction along the backbone of DNA. The spectral position is typical for B-DNA. As we can see the intensity in this line shows a variation which is coincident with the features of the white light image in Figure 3B. Dark bands in this image coincide with a high DNA signal. The immediate conclusion is that in the bands a higher DNA concentration is present. The protein distribution can be measured using the band at 1449 cm-1. This band originates in the -CH2-bending motion of aliphatic side chains in the proteins. Comparison of the spatial distribution of intensity in this band with the white light image shows that dark bands are more rich in protein than the interbands. These examples illustrate the power of Raman microscopy; a direct correlation can be made between morphology and the composition of materials.

Figure 3. A comparison of a line scan Raman image (A) of a Chironomus
polytene chromosome with the white light image (B).
On the horizontal axis in (A) the vibrational frequency is imaged
and perpendicular a spatial dimension.
The spatial dimension is chosen to run along the long axis of the chromosome.
Full spectral information is obtained together with a single spatial dimension.
The confocal direct imaging Raman spectrometer is schematically presented in Figure 4. The set up is constructed with a minimal number of optical components. Versatility in the choice of laser frequencies was desired. For that reason both an argon-ion laser as well as a krypton-ion laser were used as light sources. The laser beam was coupled into the microscope via a dichroic mirror and a bi-axial scanning mirror. The objective was filled at all scanning angles. The movement of the scanning mirror (~5 Hz and ~70 Hz in perpendicular directions) creates an illuminated area under the objective with a rectangular shape. The back-scattered light is collected by the same objective and again passed along the scanning mirror. In this way the beam of scattered light is essentially fixed in space between the scanning mirror and the detector C1. The Raman scattered light is transmitted through the dichroic mirror, additionally filtered for further laser light suppression and imaged onto a pinhole, that acts as the entrance pinhole for the poly-chromator. The pinhole allows a confocal resolution in all dimensions. We have recognised the need to have next to each other the capacity to obtain Raman spectra and Raman images. For this reason two CCD cameras were incorporated, one on each exit port. Camera C1 in Figure. 4 is used to register spectra.
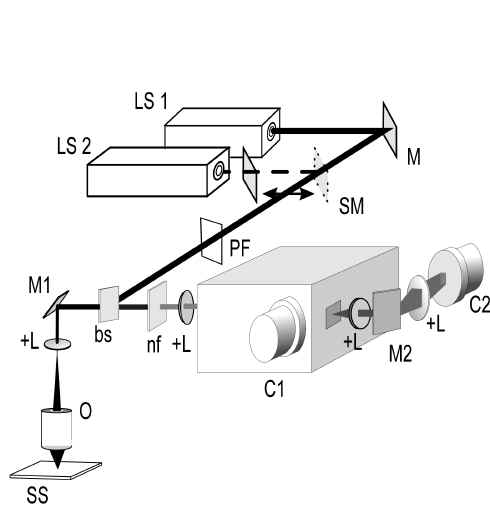
Figure 4. The Raman direct imaging spectrometer.
LS1 and LS2: Ar+– and Kr+-lasers, M: mirror, SM: sliding mirror,
PF: plasma filter, bs: beamsplitter, M1: biaxial scanning mirror
(General scanning, model no. MG325D), +L: positive lens,
O: objective, SS: sample stage, nf: notch filter,
C1: CCD camera (Princeton Instruments Inc. LN/CCD 1100 x 330 PB/VISAR)
for micro-spectroscopy, M2: biaxial scanning mirror
(General scanning, model no. MG325D),
C2: CCD camera (Princeton Instruments Inc. LN/CCD 512 TKE).
The spectrum can next be used to select a band of frequencies for imaging. The bandwidth is determined by the grating constant and the width of the exit slit, port 2. It can in this way be adapted to the bandwidth of the Raman vibration of interest and the desired spectral purity. A typical bandwidth is 10 cm-1. The light coming from port 2 is first collimated by a lens and next passes the second set of scanning mirrors (M2 in Figure 4). This set is exactly synchronised and in phase with scanning mirror set M1 (Figure 4). The amplitude of M2 can be adjusted. In this way it is possible to optimise the number of pixels in the image with the available signal-to-noise-ratio and the desired resolution.
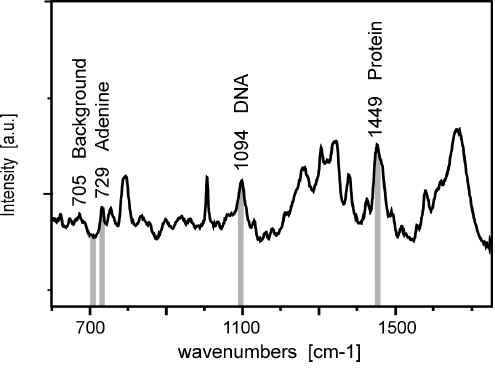
Figure 5. The Raman spectrum of a Chironomus polytene chromosome.
The assignment of bands in the spectrum is to DNA and protein components
A selection of frequency bands around for instance 729, 1094 and 1449 cm-1
enable Raman image formation respectively of adenine base residues, DNA and proteins.
In Figure 5. a Raman spectrum of a Chironomus polytene chromosome is shown. The bands in the spectrum were assigned to DNA or protein. After inspection of the spectrum images can be generated at any desired wavenumber shift. Intense bands occur at 1449 cm-1 (protein) and 1094 cm-1 (DNA). These bands are used to generate confocal Raman direct images (Figure 6). A distribution of DNA is presented in Figure 6B and of protein in Figure 6C. Also weaker bands such as the adenine vibrational band at 729 cm-1 can be used for imaging. Comparison of the DNA image in Figure 6B with the white light image in Figure 6A shows that DNA rich area’s coincide with dark bands in the chromosome. The same holds for the protein image of the chromosome in Figure 6C. Dark bands coincide with a high protein concentration. Ratio imaging of the image of the DNA – and protein distributions reveals that the protein/DNA ratio is higher in the interbands than in the bands (data not shown).

Figure 6. A comparison of the white light microscopy
(Figure. 6A) image of a chromosome from Chironomus Thummi Thummi with
(Figure 6B) the confocal Raman direct images obtained at 1094 cm-1
(DNA backbone vibration) and (Figure 6C) at 1449 cm-1 (aliphatic side chains in proteins).
Conclusions
We have constructed two Raman micro-spectrometers specifically for the research of problems in cellular biophysics. One set up employs line scanning and is only partially confocal. This Raman micro-spectrometer captures full spectral information together with one spatial dimension. The other set up enables confocal Raman micro-spectroscopy for full spectral information on one hand and imaging in selected Raman frequencies in two spatial dimensions on the other hand. The sensitivity of both set ups is good. Approximately 14% of all photons collected by the microscope objective contribute to the registered data. Low photon fluxes of a few photons per minute per pixel are sufficient to generate images of chromosomes in DNA content and protein content. Unique relationships between molecular distribution and morphology are in this way obtained.
References
[1] M. Delhaye and P. Dhamelincourt, J. Raman Spectrosc. 3, 33 (1975).
[2] G. J. Puppels, J. H. F. Olminkhof, G. M. J. Segers-Nolten, C. Otto, F. F. M. de Mul and J. Greve, Exp. Cell Research 195, 361 (1991).
[3] G. J. Puppels, F. F. M. de Mul, C. Otto, J. Greve, M. Robert-Nicoud, D. J. Arndt-Jovin and T. M. Jovin, Nature 347, 301 (1990).
[4] T. Wilson, Confocal Microscopy, ed. T. Wilson, Academic Press, London.
[5] N.M. Sijtsema, C. Otto, G.M.J. Segers-Nolten, A.J. Verhoeven and J. Greve, in print Biophys. J. (1998).
[6] C. Otto, N.M. Sijtsema and J. Greve, in print Eur. Biophysics J. (1998).
[7] C.J. de Grauw, C. Otto, J. Greve, Applied Spectroscopy 51, 1607-1612 (1997).
[8] N.M. Sijtsema, S.D. Wouters, C.J. de Grauw, C. Otto, J. Greve, Applied Spectroscopy 52, 348-355 (1998).
[9] C.J. de Grauw, J.D. de Bruijn, C. Otto, J. Greve, Cells and Materials, Vol. 6, p 57-62, (1996).
[10] C. Otto, C.J. de Grauw, J.J. Duindam, N.M. Sijtsema, J. Greve, J. Raman Spectr. 28, 143-150 (1997)
[11] S. Yu. Arzhantsev, A. Yu. Chikishev, J. Greve, N.I. Koroteev, C. Otto, N.M. Sijtsema, Sumitted to J. Raman Spectr. (1998)
[12] J.J. Duindam, G.F.J.M. Vrensen, C. Otto, J. Greve, Invest. Ophth. &Vis. Sc. 39, 94-103 (1998).
[13] C.J. de Grauw, N.M. Sijtsema, C. Otto, J. Greve, J. of Microscopy 188, 273-279 (1997).
Received 11th August 1998, accepted 17th August 1998.