Diffusion kinetics of TNT in nitrile rubber via FTIR-ATR Spectroscopy
![]()
58. Diffusion Kinetics of TNT in Nitrile Rubber via FTIR-ATR Spectroscopy
Y. S. Tung, R. Mu, A. Ueda and D. O. Henderson*
The Chemical Physics Laboratory, Department of Physics, Fisk University, Nashville, TN 37208
W. A. Curby and A. Mercado
System Development, Aviation Security Resources, Federal Aviation Administration Technical Center, Atlantic City International Airport, Atlantic City, NJ 08405
* Author to whom correspondence should be sent. E-mail:hendersn [at] dubois.fisk.edu
59. Abstract
| The diffusion of TNT in nitrile rubber at different temperatures is studied by FTIR-ATR. Based on the peak frequency and intensity of the NO2 symmetric and asymmetric stretch vibrations, the TNT diffused in nitrile rubber is in an isolated molecular state and/or cluster form. The diffusion kinetics is of the Fickian type. The diffusion coefficient of TNT in nitrile rubber is on the order of 10-7 cm2/s and the activation energy is in the range of 30 – 35 kJ/mol based on the kinetic model fitting. Key words: Diffusion, FTIR-ATR, 2,4,6-Trinitrotoluene, explosives |
60. Introduction
The transient diffusion of low molecular weight organic solids, liquids, and gases through polymers has been of great interest in materials science and polymer physics.[1-6] Additionally, the diffusion properties of energetic molecules such as 2,4,6-trinitrotoluene (TNT) through food wraps, rubber gloves etc. are very important for developing more effective explosive detection systems. Nevertheless, to the best of our knowledge, there have been no studies regarding this increasingly important issue.
The diffusion properties of solid or liquid through polymer are determined by the physical and chemical properties of both the penetrant and polymer. The barrier properties of the polymers are closely related with the chain structure and the system morphology. The chain structure may include the degree of polarity, interchain forces, tendency to crystallize and chain stiffness. The morphological properties are related to the crystallinity and free volume of the polymer.
In the present report, we have chosen TNT, a prototype explosive molecule, as a penetrant and nitrile rubber as a host: 1) to develop an FTIR-ATR based technique for characterizing molecular diffusion through polymers; 2) to characterize TNT diffusion kinetics through nitrile rubber; and 3) to obtain the depth profiling parameters and determine the possible critical thickness at which the TNT molecules can be effectively blocked.
61. Experimental
2,4,6-trinitrotoluene was purchased from Chem Service with a purity of 99.0%. The as-received TNT was further purified. The purified TNT has a melting transition at 81°C based on our thermal (differential scanning calorimetry) measurements. Pure nitrile rubber was provided by Pioneer Company. The rubber was also tested by differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) in the temperature range of 30 – 250°C. No indication of any sort of structural modification, phase transition, decomposition, or chemical reactions was observed. Therefore, the nitrile rubber is considered to be stable in this temperature range.
The experimental procedure for TNT diffusion in nitrile rubber can be summarized in the following steps: 1) Solvent selection; We intend to find a liquid medium that does not induce physical or chemical changes in the nitrile rubber. FTIR-ATR was applied to determine whether the solvent diffused into the rubber after 24 hours of soaking. If so, the solvent was rejected. The final screening confirmed that the cyclohexane is a good candidate which neither affects the rubber properties nor does it dissolve TNT molecules; 2) Preparation of TNT diffusion into rubber; a piece of 1×3 cm2 nitrile rubber was placed in the TNT heated up to the desired temperature. Then, the nitrile rubber coated with TNT was withdrawn from the liquid TNT after the desired diffusion time. 3) Removal of bulk TNT from rubber surface; In order to thoroughly remove bulk TNT from surface without modifying the surface properties of the rubber and without perturbing the TNT diffused into the rubber, the sample was first put into a beaker containing cyclohexane and a certain amount of silica powder with average particle size of 10 µm. The beaker was then placed in a sonicator. Based upon our numerous test runs, a 30 minute sonication was long enough to obtain excellent surface cleaning. After the sonication, the cleaned rubber was rinsed with cyclohexane three times and blow-dried. 4) FTIR-ATR spectrum collecting; The rubber sample was mounted onto a hemi-cylindrical single pass ATR (KRS-5) prism. Consistent pressure was applied to the sample each time so that the pressure-induced fluctuations in the spectra were minimized and the air gap presented in the rubber/prism interface was eliminated. The angle of incidence was maintained at 45° throughout the FTIR-ATR measurements. A Bomem DA3 interferometer was used for collecting the infrared spectra. Typically, 400 scans were collected at 4 cm-1 resolution. All FTIR-ATR spectra of the TNT-nitrile rubber samples were recorded at 25° and after the cleaning procedure.
62. Data Analysis
In order to obtain consistent and reliable experimental results based on the FTIR-ATR measurements, a number of factors for data and error analysis must be carefully examined. They are: 1) the effects of the penetration depth on the reflectance spectra, 2) the effect of pressure on spectral intensities and penetration depth, and 3) spectrum normalization for TNT in the polymer.
Effects of Penetration Depth. As has been discussed by many researchers[7], the penetration depth dp can be written as:
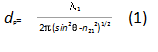
where λ1 = λ/n1, n21 = n2/n1, λ is the wavelength of the incident light, n1 and n2 are the refractive indices of the KRS-5 prism and the polymer, respectively. θ is the angle of the incidence and it was fixed at 45° throughout the present experiments. The incidence angle was well above the critical angle, so that no spectral distortion is expected. The FTIR-ATR spectra obtained from the present experiments are relatively weak and the typical peak value of the strongest band was about 0.2 absorbance units. Therefore, no saturation effects were expected or observed.
63. Pressure Effect on the Spectral Intensity and the Penetration Depth. A series of testing experiments was conducted to evaluate the effects of spectral intensity change as the function of the applied pressure with the same contact area. In order to account for intensity fluctuations arising from variations in the polymer contact with the prism, the spectra were normalized against a band at 2932 cm-1 due to CH2 stretch vibration of the polymer and the bands at 2236 and 967 cm-1 due to acrylonitrile C≡N and =C-H out-of-plane vibration from butadiene (BD),[8] respectively. Within a reasonable pressure range, the maximum spectral intensity variation was less than 2%. Therefore, spectral normalization is an effective and reliable way to overcome the pressure induced intensity variation. It is noteworthy that the choice of the relatively weak spectral bands for normalization is preferred since such bands eliminate the possibilities of saturation.
Spectrum Normalization for TNT Diffused in Nitrile Rubber. Figure 1 shows the FTIR-ATR spectra of pure nitrile rubber (1A), TNT (1B), and TNT in nitrile rubber (1C). It is clear that the band at 967 cm-1 is the unique band of the nitrile rubber. In addition, there was no severe band overlapping with any of the bands in rubber or in TNT that were used for spectral analysis. Therefore, the band at 967 cm-1 from acrylonitrile component in the nitrile rubber was chosen for spectral normalization. All of the integrated intensities reported for TNT diffusion in nitrile rubber were calculated after the spectral normalization. A similar normalization procedure has been used in literature to conduct quantitative analysis.[9]
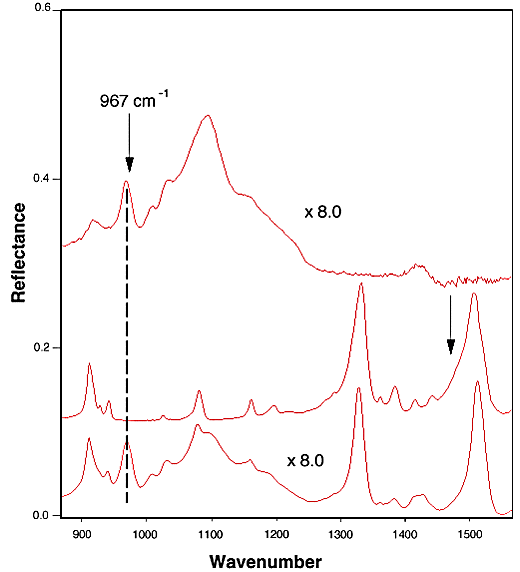
Figure 1. FTIR-ATR spectra of as-received nitrile rubber (A), TNT (B), and TNT diffused in nitrile rubber (C).
64. Results
FTIR-ATR spectra of TNT diffused into nitrile rubber at 93, 103, and 113 °C as a function of diffusion time are plotted in figures 2, 3, 4. The band at ~1542 cm-1 is attributed to the NO2 asymmetric stretching vibration of TNT molecules, while the band at ~1348 cm-1 is due to the NO2 symmetric stretching vibration of TNT.[10] Comparing the ATR peak positions of TNT in nitrile rubber at early stage with the ATR peak positions of the bulk TNT, the NO2 asymmetric stretch vibration of TNT diffused in nitrile rubber was blue-shifted ~8 cm-1 from its bulk value, while the NO2 symmetric stretch vibration was red-shifted ~2-4 cm-1. At later stage, a consistent 4 cm-1 blue-shift in NO2 asymmetric vibration was observed for TNT diffused into the rubber at 93, 103, 113°C. However, as indicated in figs. 2-4, the frequency was still 4 cm-1 higher than that of bulk TNT. Figs. 5-7 show the integrated intensity of the bands at 1542 and 1348 cm-1 as a function of the diffusion time at 93, 103, and 113°C, respectively. Clearly, within the infrared penetration depth, the amount of time for TNT to reach a saturation concentration in nitrile rubber is strongly dependent upon the diffusion temperature. At 93 °C, the diffusion results indicate that a sigmoidal type of diffusion process is operative. That is, a comparatively slow rate of diffusion is followed by a faster diffusion process at t > 150 min. At a higher temperature, however, only one step diffusion process prevailed and the diffusion reaches its saturation in a much shorter time interval. As indicated in figs 5-7, the total TNT uptake by the nitrile rubber is not sensitive to the diffusion temperature used. The diffusion rate, on the other hand, was considerably increased at higher temperatures. A prolonged dosing time for nitrile rubber in liquid TNT beyond the TNT saturation times has no effects on the FTIR-ATR spectra.
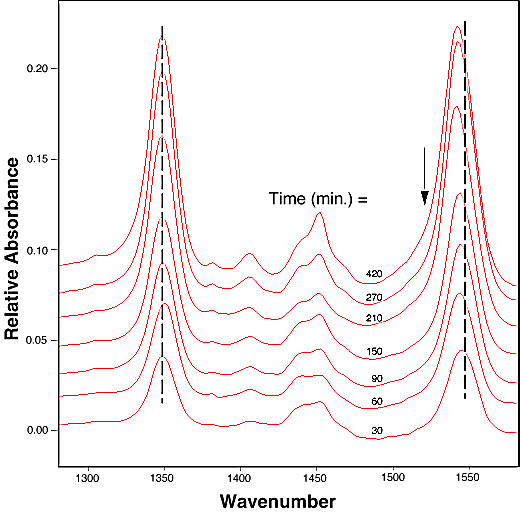
Figure 2. Infrared spectra of TNT diffusion in nitrile rubber as the function of time at 93 °C. The arrows indicate the linewidth broadening position in later stage of the dosing time.
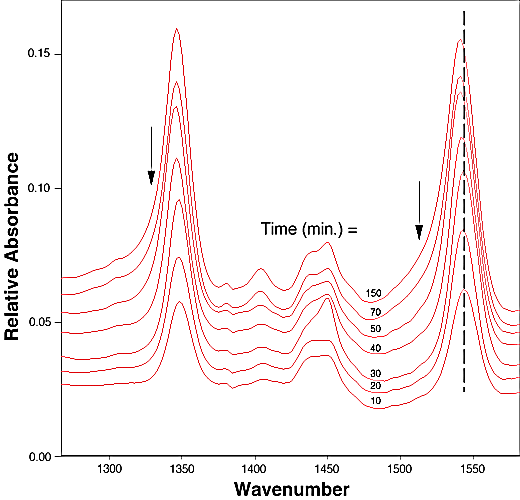
Figure 3. Infrared spectra of TNT diffusion in nitrile rubber as the function of time at 103 °C. The arrows indicate the linewidth broadening position in later stage of the dosing time.

Figure 4. Infrared spectra of TNT diffusion in nitrile rubber as the function of time at 113 °C. The arrows indicate the linewidth broadening position in later stage of the dosing time.
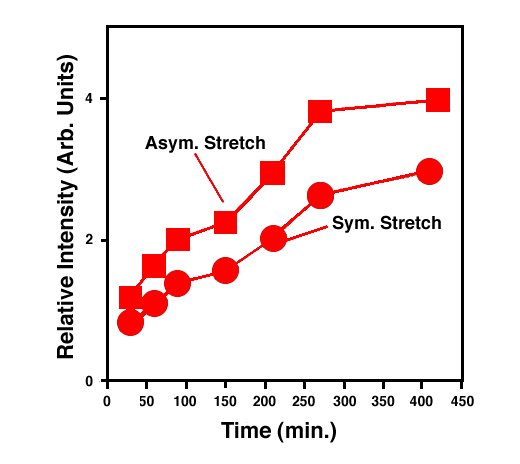
Figure 5. Integrated intensity of NO2 symmetric and asymmetric stretch vibrations of TNT in nitrile rubber at 93 °C.
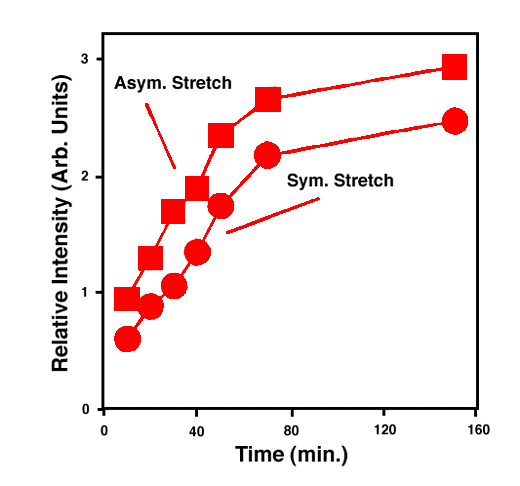
Figure 6. Integrated intensity of NO2 stretch vibrations of TNT in nitrile rubber at 103 °C.
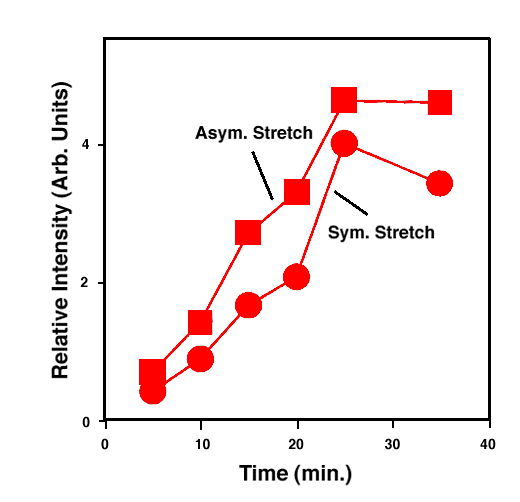
Figure 7. Integrated intensity of NO2 stretch vibration of TNT in nitrile rubber at 113 °C.
65. Discussion
TNT Molecules in NBR. The kinetics of diffusion process in polymeric material is closely related to the interactions between the diffusant and the polymer matrix [11-13] and infrared spectroscopy is an excellent tool to study the interactions. Both the symmetric and asymmetric NO2 stretching bands of TNT deserve our attentions. As we have reported previously, the TNT polycrystal NO2 symmetric stretching band is located at 1350 cm-1 and the asymmetric stretching band is located at 1533 cm-1. During the solid-liquid phase transition the peak position of the symmetric stretching band red-shifts by 2-4 cm-1 and the asymmetric stretching band blue-shifts by 6 cm-1. This opposite position shift is due to the breaking of the intermolecular hydrogen bonding between the NO2 group of the TNT molecule and the hydrogen atoms of the nearest neighbors. Hydrogen bonding is also the reason for the opposite position shift of the symmetric and asymmetric stretching of water and other systems.[14-16] The blue-shift of the peak position of the NO2 asymmetric stretch vibration of TNT diffused in nitrile rubber is 6 cm-1 more than the solid-liquid transition and the red-shift of the NO2 symmetric stretch is 2 cm-1 more than the phase transition. This larger opposite position shift of the TNT in nitrile rubber suggests that the TNT diffused into nitrile rubber is in a weaker hydrogen bonding form than TNT liquid. This weaker hydrogen bonding can be explained by considering the hydrogen bonding between TNT and the nitrile rubber- 1) hydrogen bonding between NO2 of the TNT and hydrogen of the nitrile rubber; 2) hydrogen of TNT and C≡N groups of the nitrile rubber. However, the C≡N group can form hydrogen bonding with polymer itself and present experiment can not distinguish whether C≡N form hydrogen bonding with TNT or polymer. Due to the physical size of the TNT molecule, complex distribution of the free volume in the nitrile rubber and the accessibility of the C≡N group, it is conceivable that the optimal hydrogen bonding is not formed due to the steric factors. In addition to the peak position, the linewidth can provide additional information regarding the diffusion process. At the early stage, the line width is sharp and symmetric and broadens asymmetrically at the lower frequency side with a longer diffusing time. The change of linewidth with time suggests that at the early stage of diffusion, the TNT molecule is in an isolated state and at the later stage more and more TNT diffused into the rubber and TNT formed hydrogen bonding between themselves. The increasing hydrogen bonding between the TNT molecules is also supported by a 2 cm-1 redshift in the asymmetric NO2 stretch vibration. Therefore, the diffusion kinetics at the early stage occurs predominantly by molecular diffusion and the diffusion process at the later stage is molecular diffusion coupled with possible cluster formation. It is most likely that the cluster formed inside the voids of the nitrile rubber. Due to the lack of connectivity between the voids, bulk TNT is not formed and our experimental results do not reveal solid or liquid TNT. Based on the above argument, we conclude that the diffusion kinetics of TNT in nitrile rubber sheet is best described by one dimensional molecular Fickian diffusion.
66. TNT Sorption Kinetics in Nitrile Rubber. We analyze our data based on the model proposed by Fieldson and Barbari.[17] A more general form of the equation can be expressed as At and A∞ are the reflectance (or absorbance) at diffusion time t and at sorption equilibrium. D is the diffusion coefficient and L is the half thickness of the nitrile rubber sheet in this case. Equation (2) provides a very good fit for At/A∞ ≥ 0.5 and the requirement for using equation (2) is readily met for most cases.[17]
![]()
Fig. 8 plots the data points that satisfy the condition of At/A∞ ≥ 0.5 fitted with equation (2). Both the NO2 symmetric and asymmetric stretch vibration data fit well with equation (2). The diffusion coefficient is on the order of 10-7 cm2/s, which is comparable with the diffusion coefficients of other systems.[11-13] Since the TNT molecules diffused into nitrile rubber are either in an isolated molecular state or in the cluster form, the diffusion coefficient is expected to follow Arrhenius relationship for temperature dependent molecular diffusion. That is,
![]()
where ED is the activation energy required for the penetrant (TNT) to diffuse. The fitted Arrhenius plots are depicted in Fig. 9. The activation energy was estimated to be 30 – 35 kJ/mol.
As pointed out earlier, the diffusion kinetics of TNT molecule in polymers is truly a molecular diffusion in nature. More experiments are currently underway to investigate the early diffusion kinetics for energetic molecules to diffuse in polymers.
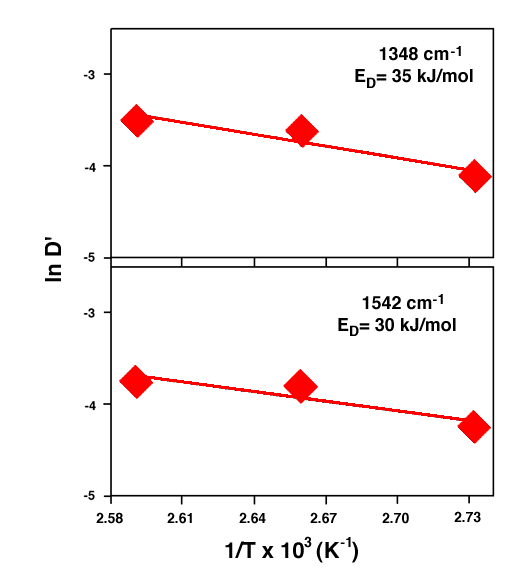
Figure 8. One dimensional Fickian diffusion model fitting from equation (3) in the text.
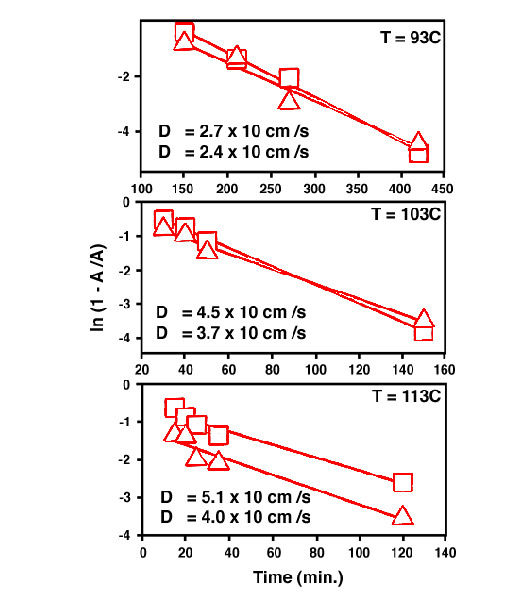
Figure 9. Arrhenious plot of TNT diffusion in nitrile rubber at 93 °, 103 °, and 113 °, respectively.
67. Conclusion
The FTIR-ATR technique has been used to study TNT diffusion in nitrile rubber at elevated temperatures. Infrared spectra suggest that TNT molecules diffused into nitrile rubber were in a molecular and or cluster state instead of solid or liquid phase. The sorption kinetics can be adequately described by Fickian diffusion. The diffusion coefficient for TNT in nitrile rubber was on the order of 10-7 cm2/s. The activation energy based on Arrhenious plot was 30 – 35 kJ/mol.
Acknowledgement
The authors gratefully acknowledge the Federal Aviation Administration for financial support under grant #93-G-057. The acknowledgment also goes to Pioneer Company for providing the nitrile rubber sample.
References
1. N. E. Schlotter, J. Phys. Chem. 94, 1692 (1989).
2. J. S. Vrentas and C. M. Vrentas, Macromolecules 26, 1277 (1993).
3. T. Ito, K. Aizawa, and J. Seta, Colloid & Polymer Sci. 269, 1224 (1991).
4. M. S. High, P. C. Painter, and M. M. Coleman, Macromolecules 25, 797 (1992).
5. J. G. Van Alsten and S. Lustig, Macromolecules 25, 5069 (1992).
6. E. Jabbari and N. A. Peppas, Macromolecules 26, 2175 (1993).
7. For example, N. J. Harrick, Internal Reflection Spectroscopy, Interscience-Wiley, New York 1997.
8. M. Sargent, J. L. Koenig, and N. L. Maecker, Appl. Spectrosc. 45, 1726 (1991) and R. A. Nyquist, Appl. Spectrosc. 41, 798 (1987).
9. S. C. Hsu, D. Lin-vien, and R. N. French, Appl. Spectrosc. 46, 225 (1992).
10. C. P. Nash, T. E. Nelson, J. J. P. Stewart, and W. R. Carper, Spectrochimic Acta 45A, 585 (1989); ibid 43A, 1249 (1987); ibid 42A, 13 (1986); 42A, 461 (1986).
11. J. S. Crank and G. S. Park, Diffusion in Polymers (Academic Press, Oxford, 1975).
12. J. S. Crank, The Mathematics of Diffusion (clerendon Press, Oxford, 1975).
13. H. B. Hopfenberg and V. Stannett, The Physics of Glassy Polymers, R. N. Haward, Ed. (Wiley, New York, 1973) Chap. 9.
14. D. Hazai and S. Bratos, The Hydrogen Bonding, ed. by P. Schuster, G. Zundel, C. Sandorfy, Vol 2, Chap. 12 pp 567 – 611 (North – Holland Publishing Company, Amsterdam, New York 1976).
15. G. C. Pimentel and C. H. Sederholm, J. Chem. Phys. 24, 639 (1956).
16. J. R. Reimers and R. O. Watts, Chem Phys. 85, 83 (1984).
17. G. T. Fieldson and T. A. Barbari, Polymer 34, 1146 (1993).
REF: Int. J. Vib. Spect., [www.irdg.org/ijvs] 1, 5, 58-67 (1998)